XiaoMi-AI文件搜索系统
World File Search Systempotent

鉴定有效的paracaspase malt1抑制剂...
结果:我们使用我们专有物理学的自由能扰动(FEP+)建模技术确定了新型的小分子MALT1抑制剂。我们的化合物显示出对MALT1酶活性的有效抑制(亚NM),以及通过表面等离子体共振(SPR)测量的MALT1蛋白的高结合亲和力(sub nm)。bcl10是MALT1的结合伙伴,在C端在C端裂解。Our inhibitors were efficacious in a target engagement assay showing prevention of BCL10 cleavage in Activated B-cell (ABC) subtype of diffuse large B cell lymphoma (DLBCL) cell lines OCI-LY3 and OCI-LY10, which are Bruton tyrosine kinase (BTK) inhibitor ibrutinib-resistant and -responsive respectively.我们的化合物是OCI-LY3和OCI-LY10细胞中IL10分泌的有效抑制剂,这与NF-κB信号传导的抑制一致。我们还检查了MALT1抑制剂对ABC-DLBCL细胞增殖的影响。我们的抑制剂在OCI-LY3和OCI-LY10细胞系中都表现出有效的抗增殖作用,以及在BTKI敏感的ABC-DLBCL细胞图中与ibrutinib的协同作用。检查蛋白酶面板和脱靶安全筛选面板以及体内高剂量耐受性研究表明,我们的化合物具有出色的选择性和明显的安全余量。等离子体IL10和肿瘤BCL10在PK/PD研究中已被鉴定为可靠的PD标记。剂量依赖性肿瘤生长抑制作用,还观察到与Venetoclax结合使用的功效。

通过表型方法发现强效 G ...
摘要:G-四链体 (G4) 序列可以折叠成更高级的 G4 结构,在人类基因组中含量丰富,并且在许多与人类癌症起始、进展和转移有关的基因的启动子区域中过度表达。它们是 G4 结合小分子的可能靶标,在启动子 G4 的情况下,会导致这些基因的转录下调。然而,目前只有极少数 G4 及其配体复合物的结构信息可用。这一限制,加上目前与大多数复杂人类癌症有关的含 G4 基因的信息有限,导致了以表型为主导的 G4 配体药物发现方法的发展。这种方法通过几代三取代和四取代萘二酰亚胺 (ND) 配体的发现得到说明,这些配体被发现在胰腺癌细胞系中表现出强大的生长抑制作用,并且在这种难以治疗的疾病的体内模型中活跃。经过多次探索,最终研发出了一种高效四取代 ND 衍生物 QN-302,目前正在进行 1 期临床试验评估。这里列出了 QN-302 下调表达的主要基因:所有基因均具有 G4 倾向,并且已发现在人类胰腺癌中上调。其中一些基因在其他人类癌症中也上调,支持了 QN-302 是一种泛 G4 药物的假设,该药物在胰腺癌之外具有潜在用途。

FDA 批准的药物硝唑尼特是一种强效抑制剂......
(未经同行评审认证)是作者/资助者。保留所有权利。未经许可不得重复使用。此预印本的版权所有者此版本于 2023 年 4 月 7 日发布。;https://doi.org/10.1101/2022.07.13.499346 doi:bioRxiv preprint
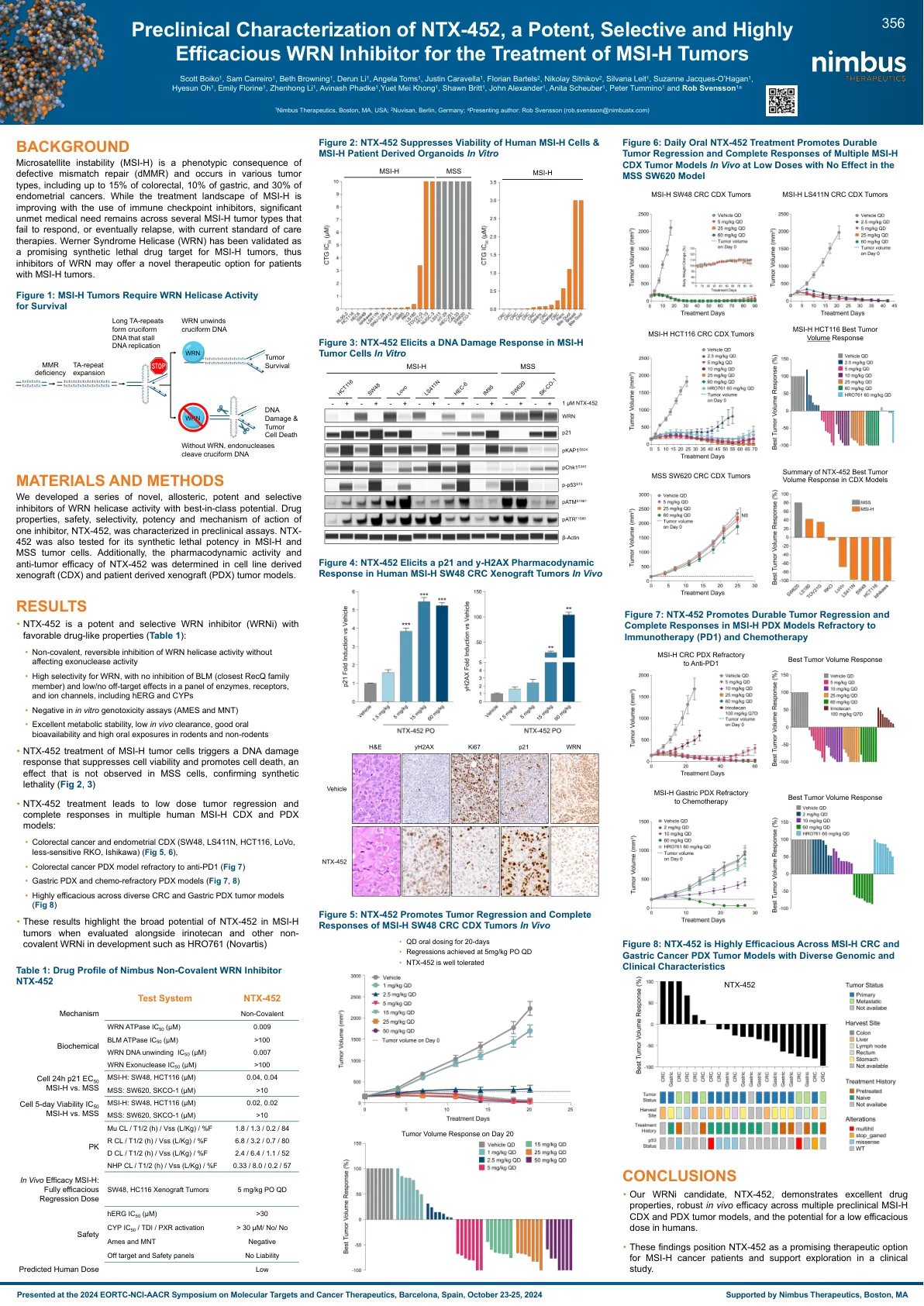
NTX-452 的临床前表征,一种强效、选择性……
微卫星不稳定性 (MSI-H) 是错配修复缺陷 (dMMR) 的表型结果,发生在各种肿瘤类型中,包括高达 15% 的结直肠癌、10% 的胃癌和 30% 的子宫内膜癌。尽管随着免疫检查点抑制剂的使用,MSI-H 的治疗前景正在改善,但对于几种使用当前标准治疗疗法无法产生反应或最终复发的 MSI-H 肿瘤类型,仍然存在大量未满足的医疗需求。沃纳综合征解旋酶 (WRN) 已被证实是 MSI-H 肿瘤的一个有前途的合成致死药物靶点,因此 WRN 抑制剂可能为 MSI-H 肿瘤患者提供一种新的治疗选择。
lorundrostat的首次人类研究,一种有效且高度...
致谢:此分析由Mineralys Therapeutics,LLC资助。早期研究由MSC Aya Fujii进行; Yuki Hiraga博士; Mizue Kawai,Bpharm;和Kanami Sugimoto-Kawabata博士;是三菱Tanabe Pharma Corporation的员工,该公司发明并超越了该化合物向Mineralys Therapeutics,Inc。编辑支持,由宾夕法尼亚州纽敦广场的Kay Square Scientific的Catherine Champagne提供。此支持由Mineralys Therapeutics,LLC资助。披露:KO是三菱Tanabe Pharma Corporation的雇员,该公司发明并超越了该大体上的Mineralys Therapeutics,LLC。DR是Mineralys Therapeutics,LLC的员工。在美国心脏病学院举行的第72届年度科学会议上与世界心脏病学大会一起于2023年3月4日至6日在美国洛杉矶的新奥尔良举行。

发现KT-474,有效,选择性和口头...
本演讲包含1995年《私人证券诉讼改革法》(PSLRA)和其他联邦证券法的含义内的前瞻性陈述。这些陈述包括有关我们当前和未来的前景以及我们的运营和财务业绩的信息,这些信息基于当前可用的信息。本演示文稿中包含的历史事实陈述以外的所有陈述,包括有关我们的战略,未来财务状况,未来经营,预计成本,前景,计划,管理目标和预期市场增长的明示或暗示陈述,都是前瞻性陈述。In some cases, you can identify forward-looking statements by terminology such as ‘‘aim,'' ‘‘anticipate,'' ‘‘assume,'' ‘‘believe,'' ‘‘coontemplate,'' ‘‘continue,'' ‘‘could,'' ‘‘design,'' ‘‘due,'' ‘‘estimate,'' ‘‘expect,'' ‘‘goal,'' ‘‘intend,'' ‘‘may,'' ‘‘objective,'' “计划,”'''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''''将''''将'和其他类似的表达方式,这些表达方式是对未来事件和未来趋势的预测或未来趋势,或这些术语的否定或其他可比性的术语或其他可比性的术语。这些前瞻性陈述包括有关我们当前和未来临床试验的启动,时间,进步和结果的陈述,以及我们的产品候选人以及我们的研究与开发计划的当前和未来临床前研究;我们计划开发和商业化当前的产品候选人以及任何未来的产品候选人,以及我们的业务模型和业务模型的实施,并为业务,当前的产品候选人和任何未来的产品候选人进行战略计划。我们实际上可能没有实现我们的前瞻性陈述中披露的计划,意图或期望,并且您不应过分依赖我们的前瞻性陈述。您不应依靠前瞻性陈述作为未来事件的预测。

基于进化的强效泛疫苗的构建
Guo 1 , Yongbing Pan 3 , Xiaoli Wu 3 , Yimin Yang 3 , Zhaofei Jing 3 , Yongzhong Jiang 4 , SARS-CoV-2 Vaccine Task Force Group 1,2 , Yu Chen 1,2 , Huan Yan 1,2 , Yu Zhou 1 , Ke Xu 1,2,* , Ke Lan 1,2,5*

垂体腺苷酸环化酶激活型过肽是有效的
1在体内研究了垂体腺苷酸环化酶激活多肽(PACAP)对微血管血流和血浆蛋白泄漏的影响。2对PACAP38(肽的38个氨基酸形式)的皮内注射,导致通过'33xe清除技术测得的血流剂量依赖性增加。每个位点PACAP38的10-2 mol诱导血流的同等增加,每个部位的人轴 - 钙蛋白基因相关肽(CGRP)和每个位点每个位点的血管活性肠肠多肽(VIP)的摩尔(VIP)10-2 mol诱导。3 PACAP38的血管扩张活性与用激光多普勒流量计测量的肽PACAP27的27个氨基酸形式无显着差异,而在每个位点10-2摩尔以上10-2摩尔以上的基础流量以上,导致104±14%,导致110±18%。4在每个位置1012 mol时,PACAP38的效果比CGRP的效果更长。在2小时,PACAP38(p <0.05)时,血流量保持在对照中的显着增加(p <0.05),而在此时,皮内CGRP后的血流恢复为对照值。5 PACAP38仅注射了对“ 25i标记白蛋白的微血管泄漏”。然而,PACAP38显着增强了缓激肽诱导的水肿,其中它比VIP高约100倍。6 divap38诱导的水肿增强并未被吲哚美辛抑制,该剂量确实抑制了蛛网膜酸抑制铁丁蛋白诱导的水肿的增强。7 PACAP38至少与其他假定在体内兔皮肤测试时所涉及的其他肽一样有效。PACAP可能有助于炎症的高度和水肿成分。关键字:垂体腺苷酸环化酶激活多肽;血管舒张;动脉;血管活性肠多肽; Cal- citonin基因相关肽;腺苷酸环化酶

进一步表征来自妊娠期间给药的双重诱发F RSV疫苗的3阶段RCT的疫苗功效
2。22。2 3开始马德里cioccc。 5 6美国加利福尼亚州圣地亚哥的辉瑞公司; 7 Pfizer Inc.,美国纽约,美国; 8美国宾夕法尼亚州Colleville Pfizer Inc.; Inc.,美国纽约,美国; 10号辉瑞公司,美国加利福尼亚州洛杉矶; 11

ASN-3186是...
24 24 24 24泛素特异性肽酶1(USP1)是DNA转移合成的关键调节剂和Fanconi贫血DNA Repition途径1,2。USP1从多种底物(PCNA,FANCI,FANCD2,PARP1,EZH2,CHK1等)中去除泛素与DNA损伤修复(DDR)3非常重要。USP1抑制剂可能会患有DDR脆弱性的某些癌症。ASN-3186是去泛素化酶USP1的选择性和有效抑制剂。ASN-3186治疗导致BRCA1/2突变的乳腺癌细胞系中的细胞死亡。ASN-3186与第一代或第二代PARP抑制剂(Olaparib/saruparib)结合使用时表现出强大的细胞杀伤协同作用。此外,ASN-3186在BRCA1/2MUT和HRD-(同源重组缺乏症)中表现出强烈的肿瘤生长抑制作用,具有主要PARPI耐药性。在头对头研究中,ASN-3186被发现比KSQ-4279(据报道的USP1抑制剂)4作为单一疗法或与Brcamut肿瘤模型中的Olaparib结合使用。正在计划进一步开发ASN-3186作为潜在的一流USP1抑制剂。