XiaoMi-AI文件搜索系统
World File Search Systemprion

一目了然
引言“ prion”一词首先是由斯坦利·普鲁瑟纳(Stanley Prusiner)于1982年创造的,以描述“蛋白质感染性颗粒”,导致各种致命和可传播的神经退行性疾病,包括scrapie,Creutzfeldt - Creutzfeldt - Jakob病(Jakob病)和Kuru(CJD)和Kuru(Prusiner,1982年)。他们的作用机理让人想起约翰·格里菲斯(John Griffith)在1967年概述的“仅蛋白质”假设中所描述的,声称存在负责刮刀的自我复制蛋白质(Griffith,1967)。一种独特的蛋白质,指定为prion蛋白(PRP),是从crapie感染的仓鼠大脑中纯化的,该仓鼠大脑对有限的蛋白酶K消化有抵抗力(Bolton等,1982; McKinley等,1983; Prusiner等,1982; Prusiner等,1982,1982,1983)。值得注意的是,PRP的浓度与感染性prion的滴度成正比,这表明PRP代表了prion的主要组成部分。基于PRP的部分序列(Prusiner等,1984),将编码该蛋白质的基因克隆在冰草感染和未感染的动物中(Oesch等,1985)。意想不到的发现PRP是由宿主基因组编码的,这表明Prion由正常细胞蛋白的改良病理形式组成。正常细胞prion蛋白(PRP C)和病理刮擦prion蛋白(PRP SC)共享相同的氨基酸序列,但主要差异在其构象和相关的生物化学特性上,例如蛋白酶抗性和溶解度(Barry等,1986; Basler等,1986; Meyer et al。,poster)(Barry等,1986; Basler等,1986; Meyer et al。)现在已广为人知的是,从PRP C到PRP SC

治疗朊病毒病的二价 siRNA
药物降低 PrP 表达对动物模型中的朊病毒病有效,目前正在进行临床测试。将 PrP 降低 50% 可延长感染朊病毒的小鼠的生存时间和健康寿命,但不能防止症状出现或阻止疾病进展。其他候选药物应寻求将 PrP 表达降低到更低的水平。二价 siRNA 是一种新型寡核苷酸药物模式,在临床前模型中具有良好的效力、耐用性和生物分布数据,这激励我们在这项技术中寻找治疗朊病毒病的新药物候选物。在这里,我们首先确定一种针对小鼠 PrP 基因的工具化合物,并确定降低 PrP 的二价 siRNA 在感染朊病毒的小鼠中的功效。然后,我们引入了含有人类 PrP 基因完整非编码序列的人源化转基因小鼠系作为识别人类序列靶向药物的工具。我们鉴定出一种针对人类 PrP 基因的高效 siRNA 序列,并确定一种包含延伸核酸和与 RNA 靶标不匹配的 3′ 反义尾的化学支架可产生更佳的效力。我们提名降低 PrP 的二价 siRNA 2439-s4 作为人类朊病毒病的新候选药物。
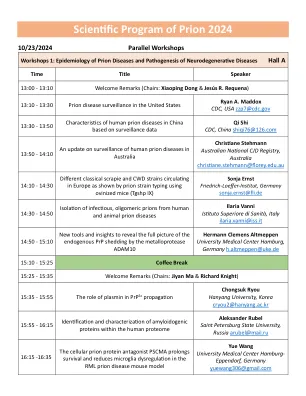
Prion 2024的科学计划
17:05-17:30皮肤中错误折叠蛋白的播种活性是神经退行性疾病的诊断生物标志物17:05-17:30皮肤中错误折叠蛋白的播种活性是神经退行性疾病的诊断生物标志物

Prion疾病中的总tau:生物标志物总tau如何促进prion疾病中的prion菌株的诊断,分类和预后
抽象的病毒疾病,也称为可传染性海绵状脑病,是一种罕见的神经退行性疾病形式,其中健康细胞prion蛋白(PRP C)的错误折叠到疾病的PRP SC形式中散布在中枢神经系统的整个结构中(CNS)。最常见的prion疾病变体是零星的克鲁特兹菲尔特 - 贾科布疾病(SCJD),但由于长期孵化时间和与其他神经退行性疾病的相似之处,诊断是具有挑战性的。脑脊液中的生物标志物总tau(T-TAU)起着重要作用,因为它是由神经退行性变化引起的浓度增加在诊断和分类的prion疾病中。该荟萃分析的目的是分析T-TAU对prion疾病诊断,分类和预后通过在RSTUDIO中采用荟萃分析方法来诊断,分类和预后。根据系统文献搜索以及对照组类型(健康,非CJD,阿尔茨海默氏病)选择的18项研究,对T-TAU敏感性和特异性的异质性和发表偏差进行了统计评估。结果表明,T-TAU的平均灵敏度为83.5%,平均特异性为86.1%。亚组围绕相似值,T-TAU在非CJD亚组中表现最好。异质性分析表明,在整个研究中,异质性中等到高异质性,漏斗图表明出版物偏差很小。总而言之,T-TAU在与其他神经退行性疾病区分开的SCJD方面表现出很高的灵敏度和特异性,但是通过结合生物标志物可以提高准确性。需要进一步的研究,以解决用T-TAU对prion菌株的分类和预后。

细胞prion蛋白的治疗靶向
摘要PRP SC是细胞prion蛋白(PRP C)的一种错误折叠的,可聚集的亚同工型,是负责人类和其他哺乳动物致命神经退行性疾病的传染性prion剂。PRP SC可以采用不同的致病构象(prion菌株),这些构型可以抵抗潜在的药物或获得耐药性,这对有效疗法的发展构成了挑战。由于PRP C是任何prion菌株的义务前体,并且是Prion神经毒性的介体,因此它代表了prion疾病的有吸引力的治疗靶标。在此MinireView中,我们简要概述了靶向PRP C的方法,并讨论了我们最近对Zn(II)-BNPYP(一种prp c -prp c -targeting卟啉粘蛋白,具有前所未有的双峰作用机制。我们认为,对Zn(ii)-BNPYP靶向PRP C可能导致新的双重机理抗prion化合物的分子机制的深入理解。关键词:抗腐蚀药物;抗PRP C抗体;反义寡核苷酸;神经变性;药理学伴侣;卟啉prion病; prp c degrader; prp c脱落;锌指抑制剂

多站点皮肤活检与脑脊液用于诊断prion疾病的prion播种活性
设计,设置和探索性队列的参与者,患者从2021年9月15日至2023年12月15日招收,每3个月进行一次跟踪,直到2024年4月。从2023年12月16日至2024年6月31日,验证性队列招募的患者。探索性队列是在Xuanwu医院的神经病学部门的一个中心进行的。验证性队列是一项涉及中国4家医院的多中心研究。参与者包括那些被诊断出患有可能零星的克鲁特兹菲尔特jakob疾病或遗传确认的PRD的人。不确定诊断或失去随访的患者被排除在外。所有患有PRD的患者在3个部位(近耳区,上臂,下背部和大腿内侧)进行皮肤采样,其中一部分同时采用了CSF样品。在确认的队列中,同时从一部分PRD患者中收集了单个皮肤活检部位和CSF样品。

●prion疾病●Guillain-Barré综合症●慢性...
多态性本身不是致病的,但是MM的纯合变体增加了SCJCH的风险(约72%的MM,VV为17%),而杂合基因型似乎具有保护性。

人类和动物中的prion病的治疗观点
prion病是一群致命的神经退行性疾病,这些疾病影响了几种哺乳动物,包括人类,牛,子宫颈,绵羊,山羊等[1-3]。这些疾病是由细胞prion蛋白(PRP C)错误折叠到β-片 - 片结构中的驱动的。疾病相关的prion称为PRP SC,易于聚集和神经毒性[4-6]。prion疾病可以与prion蛋白基因(PRNP)中的突变有关,该突变是由expo to po to to to to to to to po to to to to to to to to to to to to to to to to to to to to to po to to po to to prodion to to to to to to to po to to to po to to to prodion污染材料,也可以偶发地出现[3,7]。prion领域的研究领域是药物发现。尽管跨越了几十年的努力,但prion疾病仍然没有明显的修改治疗或治疗方法[1、2、7-10]。Several factors affect the development of effective anti-prion treatments including the different conformations that PrP Sc particles acquire (also known as prion strains [ 7 , 11 ]), the limited number of prion strains that are available to be studied in cell-culture models [ 1 ], the rare inci- dence of human prion diseases that limit patient recruitment, and the still elusive understand- ing of prion pathogenesis, among others [ 7 , 12 ].此外,缺乏在早期阶段鉴定该疾病的敏感检测方法[13]使得在脑损伤已经广泛且可能不可逆的情况下,在症状发作之前就很难治疗个体[3]。本文重点介绍用于开发针对PRP C,PRP SC和相关病理途径的抗原疗法的不同方法。我们还讨论了该领域已采用的方法来开发和评估新疗法。最后,我们讨论了在人类和动物中测试的一些治疗策略,以及针对抗王室疾病的疗法的未来观点。

对私生病治疗和预防症的新影响
prion疾病是罕见的,致命的,进行性的神经退行性疾病,会影响动物和人类。人类prion病主要作为克鲁特兹菲尔特 - 雅各布疾病(CJD)。但是,没有可治愈的疗法,动物prion疾病可能会对生态系统和人类社会产生负面影响。在过去的五十年中,科学家致力于为prion疾病找到可用的治疗或预防剂。已显示许多化合物在有关prion疾病的实验研究中有效,但毒性的局限性,不良的效应和药代动力学低。CJD最早的临床治疗方法几乎是用抗感染药物对课程改善的。随着病原性错误折叠蛋白(PRPSC)的发现并增加了对prion生物学的见解,大量新技术试图消除PRPSC。本综述介绍了有关临床和实验性prion疾病的新观点,包括免疫疗法,基因治疗,小分子药物和干细胞疗法。它进一步探讨了与这些新兴治疗方法相关的前景和挑战。

神经胶质性吞噬体系统的损害驱动prion ...
。cc-by-nc-nd 4.0国际许可证(未经同行评审证明)获得的是作者/资助者,他授予Biorxiv授予Biorxiv的许可,以永久显示预印本。它是此预印本版本的版权持有人,该版本发布于2024年2月5日。 https://doi.org/10.1101/2023.10.04.560952 doi:Biorxiv Preprint