XiaoMi-AI文件搜索系统
World File Search SystemrRNA

16S rRNA Gen Gen分析塑料破坏细菌,印度尼西亚南部苏门答腊
抽象河流是塑料进入海洋的主要途径,包括穆西河河口。能够通过聚合酶酶降解塑料废物的细菌特征。这项研究的目的是确定细菌分离株降解塑料并确定降解塑料废物的细菌的能力。这项研究使用塑料瓶,尼龙网和小吃包装器作为降解测量的对象。使用通用PCR引物对16S rRNA基因进行鉴定,以向前引物63F(5'-CAG GCC TAA CAC ATG CAA GTC-3')和反向引物1387R(5'-GGGG cgg cgg cgg cgg cgg cgg wgt gta gta caa ggcc-3')的形式进行细菌的鉴定分析。是20天内降解百分比最高的细菌类型,总计7.75%,是阿米洛里克法氏芽孢杆菌。使用16S rRNA基因分析对塑料降解细菌的类型鉴定显示了11种细菌,其中包括8种类型,包括HOMINIS葡萄球菌,铜绿假单胞菌,Acinetobacter sp。,acinetobactoctocter sp。,acinetobactobacter baumannii,baumannii,baumannii,acinetabacter abilis sp sp sp sp。淀粉法。细菌塑料降解的百分比相对较小,因此最好寻找可能有细菌生长的时间。

微生物组种类分析,用于全长16S rRNA测序的kinnex套件
结果表明,Kinnex 16S测序可以在单个Revio SMRT单元格上以1,536-plex或在续集IIE SMRT单元格上的768-plex下的平均读数> 30k平均读数。图5a显示了续集II系统上的常规全长16S库的显示2.1-330万(M)的读取,而Kinnex则显示为19.6-28.4 m,在Revio系统上,从8.5-10.1 m的Revio系统上读取,而没有Kinnex至62.5–72.5–72.2 m读取Kinnex。 此差异允许每个SMRT单元格多路复用和每个样品的读取深度更高。 将Kinnex 16S与标准FL 16S进行比较,我们发现物种组成有很高的相关性(图5B)。 在20种物种的微生物标准上,Kinnex 16S库与预期的物种表示相比,由于读取深度较高,因此与常规16S库相关(图5C)。显示2.1-330万(M)的读取,而Kinnex则显示为19.6-28.4 m,在Revio系统上,从8.5-10.1 m的Revio系统上读取,而没有Kinnex至62.5–72.5–72.2 m读取Kinnex。此差异允许每个SMRT单元格多路复用和每个样品的读取深度更高。将Kinnex 16S与标准FL 16S进行比较,我们发现物种组成有很高的相关性(图5B)。在20种物种的微生物标准上,Kinnex 16S库与预期的物种表示相比,由于读取深度较高,因此与常规16S库相关(图5C)。

改进了基于16S rRNA基因扩增子的微生物组研究的DNA提取和扩增策略。
摘要:下一代测序技术通过启用微生物组的社区级序列分析来推动人类微生物组研究的快速发展。尽管所有微生物组测序方法都取决于从样品中恢复DNA作为第一个关键步骤,但裂解方法可能是微生物组谱偏差的主要决定因素。基于温和的酶的DNA制备方法可保留DNA质量,但可以通过未能打开难以溶的细菌来偏向结果。诸如珠子跳动之类的机械方法也会偏向DNA恢复,因为打破较硬的细胞壁所需的机械能可以剪切更容易裂解的微生物的DNA,并且剪切可能会根据跳动的时间和强度而变化,从而影响重复性。我们引入了一种非机械,非酶,新型的新型快速微生物DNA提取程序,适用于16S基因基因基因的微生物组分析应用,以消除珠子的跳动。同时应用碱性,热量和洗涤剂(“快速”方案)在毫克量样品中提供了一致的在困难且易于裂解的细菌等于或更好的群体中,与现有方案相等或更好,从而产生足够的高素质DNA,用于全长长度16S RRNA基因PCR。使用包含困难和易于裂解细菌的模拟细菌群落评估了新型的“快速”方法。人类粪便样品测试将新型快速方法与标准的人类微生物组项目(HMP)方案进行了比较,该方案为肺癌患者和对照组的样品进行了比较。使用PACBIO平台上的V1V3和V4区域的V1V3和V4区域的16S rRNA基因测序分析了从两种方法中恢复的DNA。我们的发现表明,“快速”方案始终产生较高水平的公司物种,这些物种更准确地反映了细菌群落结构的特征,这通过模拟社区评估证实。新型的“快速” DNA裂解协议减少了珠子跳动和酶裂解方法常见的种群偏见,提供了改善微生物社区分析的机会,并结合了将样品输入减少到10毫克或更低的情况,并且可以启用快速传递和同时传递标准板格式中96个样品的裂解。与广泛使用的商业方法相比,这会导致样品处理时间的降低20倍,总体优势降低了2倍。我们得出结论,新型的“快速” DNA提取方案为16S rRNA基因扩增子测序的粪便提供了可靠的替代方法。
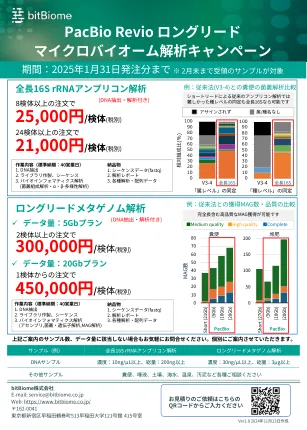
全长 16S rRNA 扩增子分析
bitBiome Inc. 电子邮件:service@bitbiome.co.jp 网站:https://www.bitbiome.co.jp/ 日本东京新宿区早稻田鹤卷町 513 号 162-0041 早稻田大学第 121 栋 415 室

Novel Variant and Known Mutation in 23S rRNA Gene of Mycoplasma pneumoniae, Northern Vietnam, 2023
ycoplasma pneumoniae is a common etiologic agent of community-acquired pneumonia (CAP) among children. Although M. pneumoniae infection often causes a mild and self-limiting dis- ease, pneumonia develops in ≈ 10%–20% of pediatric patients ( 1 ). First-line therapies for M. pneumoniae infection are based on macrolides, a group of anti- microbial drugs widely used in outpatient settings because of their high oral bioavailability. However, overuse and indiscriminate use of macrolides have contributed to the emergence of macrolide-resistant M. pneumoniae (MRMP). Point mutations in the V region of the M. pneumoniae 23S rRNA gene have been associated with macrolide resistance ( 2 ). In recent years, prevalence of MRMP has increased and is very high in Asia (13.6%–100%) ( 2 – 4 ). Dur- ing spring/summer 2023, hundreds of children with CAP were admitted daily to each of the major hospi- tals in Hanoi, Vietnam. M. pneumoniae has emerged as the major pathogen detected in approximately one third of patients with CAP (5). We analyzed the mutations in the 23S rRNA gene of M. pneumoniae isolated from nasopharyngeal samples of pediatric CAP patients during the 2023 outbreak in Vinmec Times City Hospital, Hanoi.

speciateit和vspeciateB:每序列16S rRNA基因分类学分类
图1。(a)VspeciatedB V1V3,V3V4和V4模型的十倍交叉验证表明,来自“已知物种”的序列的出色分类,模型中至少存在1个序列。“新物种”的大多数序列在某些分类级别正确分类。(b)“新物种”的查询序列的后验概率往往相对于不正确的分类而正确分类。

基于16S rRNA基因测序的肌肉减少症患者的肠道菌群的变化:系统评价和荟萃分析
肌肉减少症是一种退化性疾病,与年龄相关的骨骼肌肉质量和力量丧失,这与发生在内的不良结果有关,包括跌倒,骨折,身体残疾和死亡。在生命发展过程中,肌肉质量的损失始于40岁,每10年减少约8%,加速至70岁以后,持续到死亡(1)。肌肉减少症的患病率很高:60-70岁的个体中有5-13%,在80岁及以上的人中为11-50%(2)。多次深入研究发现,评估肌肉力量比肌肉质量更强。结果,诸如欧洲肌肉减少症工作组(EWGSOP2)和亚洲肌肉减少症工作组(AWGS)等国际肌肉减少症研究组织(AWGS)在2019年引入了新的肌肉减少症诊断的定义,重点介绍了肌肉力量(3,4)。使用标准的组合(包括低肌肉质量,肌肉力量降低和身体表现受损)建立了肌肉减少症的诊断。具体而言,使用诸如双能X射线吸收仪(DXA)或生物电性阻抗分析(BIA)等技术评估肌肉质量。低肌肉质量的阈值定义为男性<7.0 kg/m 2,女性的阈值<5.5 kg/m 2。肌肉力量通常是通过手工束强度测试评估的,男性的截止值<27 kg,女性<16 kg。通过测量步态速度(阈值<0.8 m/s)或使用短体性能(SPPB)来测量身体表现。这些标准与欧洲老年人(EWGSOP)和其他相关指南的欧洲肌肉减少症的建议保持一致(4,5)。根据EWGSOP指南,根据低肌肉质量和低肌肉功能(力量或表现)诊断肌肉减少症。肌肉减少症的阶段进一步分类为前麻痹(单独的肌肉质量低),肌肉减少症(低肌肉质量和低肌肉力量或表现低)和严重的肌肉减少症(低肌肉质量,低肌肉力量,低肌肉力量和低身体表现)。然而,由于肌肉减少症的复杂病理生理学涉及多种相关途径和有限的理解,目前缺乏临床治疗中的单一有效靶向治疗药物。

使用16S rRNA和宏基因组续集的研究
番茄果实成熟是由关键基因的脱甲基触发的,这会改变其转录水平,从而启动和传播一系列的生理事件。未知的是,当使用后票后实践成熟水果以扩展保质期时,这些过程如何改变,因为这些实践通常会降低水果的质量。为了解决这个问题,评估了处理后处理诱导的果实DNA甲基甲基和转录组的变化,以及它们如何与成熟速度相关,并评估了乙烯,脱甲酸和类胡萝卜素等成熟指标。这项研究通过动态分子变化全面连接生理事件的变化。成熟的果实在20℃,12.5℃或5℃冷却后达到“转动”(t),将其与新鲜的水果“ FHT”进行了比较。储存在12.5℃的水果具有最大的表观遗传标记和基因表达的改变,超过了后冷却后引起的变化。果实生理和年代年龄在12.5℃下取消耦合,因为成熟时间是最长的。成熟到12.5℃的果实成熟并不是最新的。没有呼吸道或乙烯爆发,而是脱落酸含量很高。在甲基化组和转录组中明显明显的后脱水和“ FHT”之间的明显差异。在“ FHT”果实中光合基因和叶绿素水平的较高表达表现为光明,因为它影响了果实成熟的分子变化。最后,对由DNA甲基化调节的基因的 - 组数据的相关分析。总体而言,这些数据改善了我们对番茄果实成熟方式如何通过后票后实践改变的解释,并且期望长期有助于提高水果质量。

16S rRNA基因的基于不同鸟类粪便的微生物群在很大程度上独立于DNA保存和提取方法
鸟类肠道菌群一直是最近关注的主题,对诸如家禽行业,微生物生态学和保护等各种领域的潜在影响。粪便微生物群经常用作肠道菌群的非侵入性代理,但是从鸟类粪便中提取高质量的微生物DNA经常被证明具有挑战性。在这里,我们旨在评估两种DNA保存方法(95%乙醇和rnalater)和五种提取方法(Indispin病原体试剂盒,Qiaamp PowerFecal ProFecal Pro DNA Kit,Microgem Prepgem Prepgem细菌试剂盒,Zymbobiommics DNA Miniprep Kit和In Bane In In Base AVA AVA)用于研究。对这些方法对来自最初三种禽类(鸡,鸵鸟和无飞行parrotkākāpō)的粪便样品的功效的系统测试发现,提取的DNA的质量,数量和完整性在提取的DNA的质量,数量和完整性上存在实质性差异,但对16S rRNA Gene基于基于基于的RRNA Gene Microbobiota profiles的质量,数量和完整性却微不足道。随后在10种系统发育和生态上多样化的鸟类物种上选择了保存和提取方法的选定组合,重申了所选方法的疗效,细菌群落结构通过技术复制的特定禽类强烈聚集。我们发现,提取功效的明显差异似乎不会影响16S基因基因的细菌群体概况,为正在进行的对鸟类肠道微生物群的研究奠定了重要的基础。

基于16S rRNA测序的阿尔茨海默氏病谱系患者的肠道菌群变化:系统评价和荟萃分析
方法:PubMed的系统和全面文献搜索,Web of Science,Embase,Cochrane图书馆,中国国家知识基础设施,中国生物学医学医学盘数据库,Wanfang数据库和社会科学引文指数数据库是从Incepper进行的,直到2023年1月。包含和排除标准,两名研究人员独立筛选并从选定的研究中提取了信息。根据“ Cochrane System评估器手册”评估了数据质量,并使用具有标准化平均差异(SMD)的Stata 14软件和95%的置信区间(95%CIS)对汇总数据进行了全面分析,用于衡量效应量。此外,如果有足够的研究报告的结果,则根据亚组荟萃分析检查了地理异质性效应(与队列有关)对GM丰度的研究。最后,使用漏斗图评估了出版偏见。