XiaoMi-AI文件搜索系统
World File Search Systemsd

1 Methacton SD感应计划(第49章)| 2025-2028
名称委员会职位由塔拉·里奇(Tara Ricci)裁定/任命为持续改善管理员管理人员管理人员梅利莎·戈尔拉(Melissa Gorla Jennifer Heusser教师教师Tracy Hanson支持员工其他老师Stephanie Sawyer教师管理员老师Deborah Wittenberg老师老师Shab Maffei教师教育专家

给予大脑体育馆和游戏疗法对增加学生学习级别SD SD的集中度的影响SD Negeri Tinom
Hijratunnor,Muhamad Ali Jafar,Ummy A'isyah Nurhayati研究计划理学学士学位,健康科学学院,大学'Aisyiyah Yogyakarta *电子邮件:nhijratun@gmail.com*; jafalali48789@gmail.com*; aisyahphysio@unisayogya.ac.id抽象学习成功受到专注于研究对象的能力的影响。集中度是实现学习成功的重要方面。提供大脑体育馆和游戏疗法练习可以增加学习的集中度,因为大脑通过运动刺激,以使注意力和学习浓度的集中度增加。这项研究的目的是确定脑健身房和游戏疗法对增加V级SD学生学习集中的影响。该研究方法是一项准测试和测试后两组设计的准实验研究,使用随机抽样进行抽样技术。研究样本的V级SD学生的样本,由30名学生组成的样本,由15组I和15组II组组成,每周进行3次,持续4周。陆军阿尔法测试用作测量浓度的测量工具。配对样品T检验组I P = 0,000(P <0.05)和II组P = 0,000(P <0.05)的结果表明,这两种练习都会影响学习浓度的改善和独立样品t检验的结果,指示P = 0.98(P> 0.05),这意味着在大脑和娱乐浓度上没有差异。进一步的研究人员的建议,提供脑健身房和游戏疗法练习以及干预措施或其他变量。简介Kesimpulan,Terdapat Peningkatan Konsentrasi Belajar Setelah Pemberian pemberian Latihan Brain Gym Dan Play Therapy,Tetapi Tidak Ada Perbedikan Antara Antara Brain Antara Brain Gym dan Play play Play疗法Terhadap Peningkatan Konsentrasi Belajar。Kata Kunci:陆军Alpha测试;脑健身房; Konsentrasi Belajar; Play Therapy脑健身房和游戏疗法对学习集中度的影响对SDN Tinom抽象学习成功的V级学生的影响受到将注意力集中在研究对象上的能力的影响。 集中度是实现学习成功的重要方面。 提供脑健身锻炼和运动疗法可以增加学习浓度,因为大脑通过运动刺激,以使学习水平和集中度增加。 该研究旨在确定脑健身房和游戏疗法对增加五年级小学生的学习浓度的影响。 这项研究的方法是一项准实验研究,并进行了测试前和测试后两组设计。 采样技术使用了随机采样。 五年级小学生的研究样本,样本量为30名学生,其中15人组成的第I组和II组15人的研究样本每周进行3次,持续4周。 陆军阿尔法测试用作测量浓度的测量工具。 结论是,在进行大脑体育馆和游戏疗法练习后,学习浓度有所提高,但是在增加学习浓度方面,大脑体育馆和游戏疗法之间没有显着差异。 关键词:陆军Alpha测试;脑健身房;玩疗法;研究浓度1。Kata Kunci:陆军Alpha测试;脑健身房; Konsentrasi Belajar; Play Therapy脑健身房和游戏疗法对学习集中度的影响对SDN Tinom抽象学习成功的V级学生的影响受到将注意力集中在研究对象上的能力的影响。集中度是实现学习成功的重要方面。提供脑健身锻炼和运动疗法可以增加学习浓度,因为大脑通过运动刺激,以使学习水平和集中度增加。该研究旨在确定脑健身房和游戏疗法对增加五年级小学生的学习浓度的影响。这项研究的方法是一项准实验研究,并进行了测试前和测试后两组设计。采样技术使用了随机采样。五年级小学生的研究样本,样本量为30名学生,其中15人组成的第I组和II组15人的研究样本每周进行3次,持续4周。陆军阿尔法测试用作测量浓度的测量工具。结论是,在进行大脑体育馆和游戏疗法练习后,学习浓度有所提高,但是在增加学习浓度方面,大脑体育馆和游戏疗法之间没有显着差异。关键词:陆军Alpha测试;脑健身房;玩疗法;研究浓度1。I组P = 0.000(p <0.05)和II组P = 0.000(P <0.05)的配对样品t检验的结果表明,这两种练习都对增加学习浓度和独立样品t检验的结果有影响,并且p = 0.98(p> 0.05)(p> 0.05),这意味着在大脑体育馆和娱乐疗法上的影响没有差异。对未来的研究人员的建议应提供脑健身锻炼,并结合其他干预措施或变量。

产品数据表 - 工业级 MICRO SD 存储卡 - Ciiva
6.1 电气描述 ................................................................................................................................................................................ 8 6.2 直流特性 .......................................................................................................................................................................... 9 6.3 信号负载 ...................................................................................................................................................................... 10 6.4 交流特性 ...................................................................................................................................................................... 11

SD 立法机关众议院司法议程 2025 年 2 月 10 日 10:00:...
一个人可以提交请求,从远程站点出席并向该委员会提供证词。请在预定会议前至少 24 小时向 housejudiciary@sdlegislature.gov 提交请求、数字讲义或书面证词。根据《美国残疾人法案》,需要帮助的个人应在会议召开前 48 小时联系立法研究委员会 (605-773-3251) 以做出任何必要的安排。

2024 年度报告和 2025 年州水资源计划 - SD DENR
danr.sd.gov 州长克里斯蒂·诺姆和第一百届立法会议成员 根据州法律的要求,随函附上水利和自然资源委员会 (以下简称“委员会”) 的 2024 年度报告/2025 年州水资源计划。 年度报告介绍了过去一年的水资源开发和废物管理活动。州水资源计划概述了州水利设施计划和州水资源管理系统 (SWRMS) 中的项目。 通过本文件,您将看到全州对水、废水和固体废物项目的持续需求,以及州政府援助对于建设这些项目的重要性。 在过去的一年里,委员会为市政饮用水、废水、农村用水、流域恢复、固体废物处理和回收项目的规划、设计和建设提供了超过 4.711 亿美元的赠款和贷款资金。2025 年州水利设施计划目前包括 61 个尚未获得资金资助的项目,预计州政府资金需求接近 5.302 亿美元。农业和自然资源部真诚感谢所有为州水资源计划的成功做出贡献的人的关注和帮助。该部门将继续与州长、立法机构、水资源和自然资源委员会以及当地项目赞助商合作,继续将州水资源计划作为通往更健康的南达科他州的路线图。诚挚的,

2024州水计划-SD DENR-南达科他州
2024年州水计划概述1972年州立法机关制定了州水计划,以确保该州水资源对一般健康,福利,安全和经济福祉的最佳总体利益,通过保护,发展,管理,管理,管理和使用这些资源。立法机关通过水与自然资源委员会(董事会)负责该计划的责任。SDCL 46A-1-2中建立的国家水计划由两个组成部分组成:国家水设施计划和州水资源管理系统。要考虑到州供水计划,项目必须符合董事会建立的标准。在考虑包含在州供水设施计划的项目时,董事会和农业和自然资源部(部门)将这些资格标准用作指南。在州立法机关中只能对州水资源管理系统的增加或删除。国家供水设施计划国家供水计划(设施计划)是潜在水项目的清单。设施计划包括农村,市政和工业供水,废水收集和处理设施,雨水污水处理机构,地下水保护和流域修复等项目。董事会负责批准将项目安置在设施计划中。董事会可以为计划的项目提供直接帮助,并在计划上安置可能会影响联邦和其他州机构的资助决定。在2023年11月,董事会考虑了43个要求在州供水计划上安置的申请。董事会在设施计划中放置了42个项目,将2024个州水设施计划的项目总数提高到390(表12和表13)。建议在州水资源管理系统列表中列出一个项目,请参见附录B。表12中的项目已获得部分或全部资金。已从董事会获得资金的项目仍在设施计划中,直到项目完成为止,并有资格要求额外资金。截至2023年12月31日,表13中的项目或项目的一部分尚未获得资金。在2022年11月在计划期间进行的计划上放置的项目,该计划在2023年日历年期间保留在2024年12月的设施计划中。2023年11月,该计划持续到2025年12月的计划。

SD立法机关司法部会分钟2/10/2025 10:00:...SD立法机关司法部会分钟2/10/2025 10:00:...
Roll Call Present: Rep. Mortenson, Rep. Reisch, Rep. Walburg, Rep. Roby, Rep. Fitzgerald, Rep. Reimer, Rep. Hughes, Rep. Massie, Rep. Kull, Rep. Pourier, Rep. Hunt, Rep. Soye, and Rep. Stevens The meeting was called to order by Representative Stevens MOTION: TO APPROVE THE MINUTES OF FRIDAY, FEBRUARY 07 TH Moved by: Walburg Second by:大豆行动:由语音投票HB 1128盛行:修改与16岁以下儿童有关的某些规定。提出:代表艾琳·希利(Erin Heaily)动议:到表HB 1128移动者:大豆第二:菲茨杰拉德(Fitzgerald)行动:由多数成员选举(13-0-0-0-0-0)选举是:承担公民逮捕或协助合法逮捕的下班执法人员。提出者:代表Dylan Jordan反对者:南达科他州警长协会的Staci Ackerman,尤里卡·詹娜·塞弗里恩,南达科他州警察局长协会,皮埃尔·史蒂夫·史蒂夫·西格尔,南达科他州南达科他州审判律师协会,皮埃尔·希伯特(Piaim re re):由多数成员选举(12-1-0-0)投票的盛行是:莫滕森,瑞奇,沃尔堡,罗比,菲茨杰拉德,菲茨杰拉德,雷默,休斯,休斯,马西,库尔,库尔,大豆,大豆和史蒂文斯投票:亨特·HB 1214:为宗教组织的安全人员提供免疫力。提出:代表Dylan Jordan支持者:代表Phil Jensen

SD员工健康计划的状态FY25生物识别筛查表格
1。填写所有参与者信息,包括电子邮件地址,并签署表格。2。在4/2/2024和4/1/2025之间访问您的初级保健提供者,参加您的年度健康考试。要求您的提供商完成生物识别筛查信息部分并签署表格。3。与您的提供商进行审查结果。4。必须在4/1/2025之前收到表格。通过上传https://www.totalwellnesshealth.com/gravity-landing/sdpf/(首选方法)或传真到402-939-0931,仅提交您的表格一次。截止日期后收到的表格将不接受。5。在提交表单后的48小时内,将发送确认电子邮件发送到您提供的电子邮件地址。如果在48小时内未收到确认电子邮件,请重新提交您的表格。将处理4/2/2024 - 6/30/2024之间提交的表格,并在7/1/24发送确认通知。6。请允许10个工作日,以便您的信息在您的LiveWellsd帐户中可用。7。预防性护理,例如健康计划的年度健康检查,涵盖了健康计划。但是,在年度健康考试期间,如果确定了单独的诊断或关注点,并且需要进行其他检查,则这些检查将以正常的计划福利支付,但可免赔额和/或共付额。参与者信息(由参与者完成)

L3harris Technologies INC DE 的 SD 表格已于 2024 年 5 月 28 日提交
我们是国防工业领域值得信赖的颠覆者。我们以客户的关键任务需求为中心,提供连接太空、空中、陆地、海洋和网络领域的端到端技术解决方案。我们为 100 多个国家的政府客户提供支持,其中最大的客户是美国政府的各个部门和机构及其总承包商。我们的产品和服务既有国防和民用政府应用,也有商业应用。我们通常直接向客户销售,并利用代理商和中介销售和营销一些产品和服务,尤其是在国际市场上。我们的运营结构主要围绕我们销售的产品、系统和服务以及我们服务的市场,并在以下四个可报告部分报告了截至 2023 年 12 月 29 日的财政年度的财务业绩:
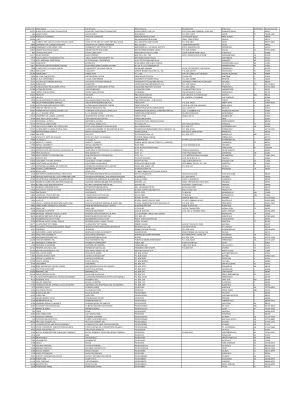
供应商 ID 订单名称 汇款名称 汇款... - Plainfield SD 202
14551 倡导慈善基金会 倡导慈善基金会 年轻之心关爱生命 3075 HIGHLAND PARKWAY, SUITE 600 DOWNERS GRIVE IL 60515 965 HEALTH EDCO HEALTH EDCO WRS GROUP P.O.BOX 21207 WACO TX 76702-1207 15366 WINSTON, STEPHANIE WINSTON, STEPHANIE WINSTON PARTY OF ONE 253 KROTIAK ROAD PARK FOREST IL 60466 10944 I.I.R.C.I.I.R.C.威廉·莫纳特大厦 148 N. THIRD STREET DEKALB IL 60115 410 威尔县巡回法院书记员 威尔县巡回法院书记员 威尔县法院 P.O.BOX 2801 JOLIET IL 60499-2801 8846 伊利诺伊大学分校 伊利诺伊大学分校 威尔县 100 MANHATTAN RD JOLIET IL 60433 12820 社区单位区 201 社区单位区 201 WESTMONT H.S.C/O KARLA GUYTON 909 N. OAKWOOD DR. WESTMONT IL 60559 13017 ARDOR HEALTH SOLUTIONS ARDOR HEALTH SOLUTIONS WELLS FARGO BUSINESS CREDIT P.O.BOX 203436 DALLAS TX 75320-3436 843 SHOOK'S PRO SHOP SHOOK'S PRO SHOP WEDGEWOOD GOLF COURSE 16534 HIDDEN RIVER CIRCLE PLAINFIELD IL 60586 14177 MUSIC PRODIGY MUSIC PRODIGY WAY OF H, INC., THE 4335 VAN NUYS BLVD STE 308 SHERMAN OAKS CA 91403 5979 VISUAL IMAGE PHOTOGRAPHY INC.VISUAL IMAGE PHOTOGRAPHY INC. W63 N582 HANOVER AVE. CEDARBURG WI 53012 7719 DCDT 区域会议 DCDT 区域会议 W167 S7752 PARKLAND DRIVE MUSKEGO WI 53150 13602 EVERWHITE EVERWHITE W158 N9332 NOR-X-WAY AVENUE MENOMONEE FALLS WI 53051 8821 阻力带训练系统 阻力带训练系统 W. 161 N 11115 MEADOW DR GERMANTOWN WI 53022 1058 伊利诺伊州公共卫生部 伊利诺伊州公共卫生部视觉和听觉 535 W. JEFFEROSN ST. 3 楼 SPRINGFIELD IL 62702 13812 FAN CLOTH FAN CLOTH VARSITY BRANDS HOLDING CO., INC. P.O.BOX 200577 ARLINGTON TX 76006 14199 TUMBL TRAK TUMBL TRAK V.T.L.INC. 5747 W. ISABELLA RD.(W M20) MOUNT PLEASANT MI 48804 4694 准时出版 准时出版 URSA MAJOR DIVISION PO BOX 639 MACOMB IL 61455 6680 C.C.B.D.会议 C.C.B.D.北德克萨斯大学会议 P.O.BOX 310860 DENTON TX 76203 5537 UIC 职业服务办公室 UIC 职业服务办公室 伊利诺伊大学@芝加哥分校 1200 WEST HARRISION,ROOM 3050 CHICAGO IL 60607 12629 NUMOTION NUMOTION 联合座椅和移动有限公司。P.O.BOX 790051 ST. LOUIS MO 63179-0051 11278 伊利诺伊州财政部办公室 伊利诺伊州财政部办公室 无人认领财产部门 P.O.I.A.S.P.A.P.O.阿灵顿高地 IL 60005 5911 S.I.L.C.地区 5 RCEP S.I.L.C.P.O.BOX 2277 BURNSVILLE MN 55337 2146 伊利诺伊州部门BOX 19496 SPRINGFIELD IL 62794-9496 15188 ULTIMATESLP.COM LORI KLEINDIENST ULTIMATESLP.COM 222 MAIN STREET #213 FARMINGTON CT 06032 3504 CENTER PUBLICATIONS CENTER PUBLICATIONS UIS BURSAR ONE UNIVERSITY PLAZA SPRINGFIELD IL 62703 10022 FROGUTS FROGUTS TWO UNION SQUARE 601 UNION ST. SUITE 4200-202 SEATTLE WA 98101 5951 I.A.S.P.A.TWO STEVENSON DR. DR. KIMBERLY CHAMBERS LINCOLNSHIRE IL 60069 3929 GALLAGHER BENEFIT SERVICES, INC. GALLAGHER BENEFIT SERVICES, INC. TWO PIERCE PLACE - 14TH FLOOR ITASCA IL 60143 499 信用管理服务信用管理服务信托部BOX 14010 SANTA ROSA CA 95402-6010 10183 自闭症家庭资源中心 自闭症家庭资源中心 TRINITY SERVICES 13318 W. LINCOLN HIGHWAY NEW LENOX IL 60451 14569 TREETOP PRODUCTS, INC. TREETOP PRODUCTS, INC. TREETOP PRODUCTS CONSOLIDATED 222 E. STATE ST. BATAVIA IL 60510 14597 乡镇高中区 214 乡镇高中区 214 交通部/ C/O AMY MCPARTLIN 2121 S. GOEBBERT RD.地区 5 RCEP 过渡会议 SIU 邮政编码 6703 CARBONDALE IL 62901 13172 伊利诺伊大学 - 运动伊利诺伊大学 - 运动售票处 1800 S. 1ST ST.香槟 IL 61820 4744 卡里格罗夫高中 卡里格罗夫高中 三橡树路 卡里 IL 60013 11512 WATKINS JR.,ELTON J. WATKINS JR.,ELTON J.THE ART OF COOL 2484 河脊 蒙哥马利 IL 60538 14137 TFH 特殊需求玩具 TFH 特殊需求玩具 TFH (USA) LTD. 4537 吉布森尼亚路 吉布森尼亚 PA 15044 2134 教育工作者员工发展 教育工作者员工发展 十沙龙路 P.O.BOX 577 PETERSBOROUGH NH 03458 12357 伊利诺伊州创新与科技部 伊利诺伊州创新与科技部 技术管理 REV 基金/DOIT A.R.BOX 10191 SPRINGFIELD IL 62791-0191 2409 WHEATON PARK DISTRICT WHEATON PARK DISTRICT TEAMS COURSE 666 SOUTH MAIN STREET WHEATON IL 60187 13845 SNO SITES SNO SITES TBP PRODUCTIONS, LLP P.O.伊利诺伊州税收部门收入税处理中心 SPRINGFIELD IL 62719 4603 I.H.S.S.B.C.A.I.H.S.S.B.C.A.STE.300 MARIETTA GA 30067 9357 APLINGTON, KAUFMAN, MC CLINTOCK, APLINGTON, KAUFMAN, MC CLINTOCK STEELE AND BARRY LTD. P.O.BOX 517 LA SALLE IL 61301 5908 INSTINCT CORPORATION INSTINCT CORPORATION STAR ROUTE BOX 78 WOODSIDE CA 94062 14604 BOLEK,JONI BOLEK,JONI ST. MARY IMMACULATE PARISH SCHOOL 。。。1625 北郊特殊教育 北郊特殊教育 特殊教育 760 RED OAK LANE HIGHLAND PARK IL 60035 1080 IMAGING PARTNERS INC. HEALTHPARTNERS AME IMAGING PARTNERS INC. HEALTHPARTNERS AME SLOT 302175 P.O.S.A.S.E.D.SUSAN MARTIN 43W432 OLD OAKS ROAD BLOOMINGTON IL 60554 10700 PARROTT, THOMAS PARROTT, THOMAS SUNRISE MUSIC PRODUCTIONS 111 S. INDEPEN LAKEWOOD CO 80232 5598 德保罗大学 DEPAUL UNIVERSITY SUITE 9500 1 EAST JACKSON BLVD CHICAGO IL 60604 15455 INGENUITY WORKS INGENUITY WORKS SUITE 407 325 HOWE STREET VANCOUVER BC CANADA 4023 ASSOCIATES IN PROFESSIONAL COUNSELING & ASSOCIATES IN PROFESSIONAL COUNSELING & SUITE 203 7300 COLLEGE DRIVE PALOS HEIGHTS IL 60463 2596 NEW ADVOCATE NEW ADVOCATE SUITE 12 1502 PROVIDENCE HWY NORWOOD MA 02062 2564 GRACE PRODUCTS CORPORATION GRACE PRODUCTS CORPORATION SUITE 111 1771 INTERNATIONAL PARKWAY RICHARDSON TX 75081-1831 12531 DISTRICT 308 DISTRICT 308 STUDENT SERVICES 4175 STATE ROUTE 71 OSWEGO IL 60543 13844 COSTUMER,THE ERIK JOHNSEN COSTUMER,THE ERIK JOHNSEN STORY FIRST INC. 1020-1030 BARRETT ST.斯克内克塔迪 NY 12305 14698 功能性生活技能 功能性生活技能刺激出版物 2470 WINDY HILL RD。BOX 66973 芝加哥 IL 60666 14765 COUNTRYVIEW LANDSCAPE & LAWNCARE, INC. COUNTRYVIEW LANDSCAPE & LAWNCARE, INC. SLAGLE, CARRIE 14632 S. KEARNS DR. 普兰菲尔德 IL 60544 13447 ASCENTIA COUNSELING AND PSYCHOLOGICAL ASCENTIA COUNSELING AND PSYCHOLOGICAL SERVICES, LLC 180 N. STETSON AVE. SUITE 3500 芝加哥 IL 60601 8639 可靠食品机械维修 可靠食品机械维修服务公司 9630 WEST 194TH PLACE 莫肯纳 IL 60448 2457 工伤赔偿 工伤赔偿自保信托 6376 EAGLE WAY 芝加哥 IL 60678 11658 创新座椅系统 创新座椅系统 座椅检查服务 8941 JEANIE LANE 法兰克福 IL 60423 12965 达科电子 达科电子 SDS-12-2222 PO BOX 86 明尼阿波利斯 MN 55486 5856景观结构公司 景观结构公司 SDS 12-0395 PO BOX 86 MINNEAPOLIS MN 55486 12317 伊利诺伊州立大学* 伊利诺伊州立大学* KNR 学校校园 BOX 5120 NORMAL IL 61790-5120 1943 S.A.S.E.D.特殊教育学校协会 6 S 331 CORNWALL ROAD NAPERVILLE IL 60540-3699 9594 学校领导简报和音频 JRNL 学校领导简报和音频 JRNL 学校管理出版有限责任公司 P.O.BOX 7105 PRINCETON NJ 08543-7105 9892 三河福音教会 三河福音教会奖学金基金 23901 W. ROLF RD.PLAINFIELD IL 60586 14139 SWIVL, INC. SWIVL, INC. SATARII INC. 1450 EL CAMINO REAL MENLO PARK CA 94025 1458 MEADOW EQUIPMENT MEADOW EQUIPMENT SALES & SERVICES 27 W. 021 ST CHARLES ROAD CAROL STREAM IL 60188-1996 13940 国家教育剧院& 艺术国家教育剧院。128 室 哥伦布 OH 43212 1630 西北互助 西北互助 RE:5145 P.O.BOX 3181 密尔沃基 WI 53201-3181 12198 TEACHER SYNERGY INC。& ARTS S. A. RAPAPORT 1400 S JOYCE ST# 832 ARLINGTON VA 22202 8674 MORROW BROTHERS FORD INC MORROW BROTHERS FORD INC RR 2 BOX 120 GREENFIELD IL 62044-9626 2387 WAUBONSEE ATHLETIC FUND WAUBONSEE ATHLETIC FUND ROUTE 47 @ HARTER ROAD SUGAR GROVE IL 60544 3290 NATIONAL AUDIO -VISUAL SUPPLY NATIONAL AUDIO -VISUAL SUPPLY ROUTE 121 EAST GRAFTON VT 05146 13577 ILLINOIS PREP TOP TIMING ILLINOIS PREP TOP TIMING ROBERT GEIGER 1074 W. TAYLOR ST. #318 CHICAGO IL 60607 13699 保险项目经理集团保险项目经理集团 RH WINE & CO., INC 225 SMITH ROAD ST. CHARLES IL 60174 1007 HORIZONS HORIZONS 康复服务 P.O.BOX 6426 BLOOMINGDALE IL 60108-6426 1622 北伊利诺伊大学北伊利诺伊大学注册 SWEN PARSON HALL ROOM 140 DEKALB IL 60115 1657 俄亥俄州立大学俄亥俄州立大学阅读康复项目 807 KINNEAR RD.TEACHER SYNERGY INC. 采购订单部门 P.O.BOX 81319 SPRINGFIELD MA 01138-1319 811 社区之友 社区之友公共艺术 310 N OTTAWA ST. JOLIET IL 60432 1703 学习伙伴 学习伙伴计划公司 1065 H BAY BLVD CHULA VISTA CA 91911 13722 PREMIER ATHLETICS, INC. PREMIER ATHLETICS, INC.PREMIERE CHEER CAMPS P.O.BOX 967 MURRAY KY 42071 10239 BEST PRICED PRODUCTS, INC. BEST PRICED PRODUCTS, INC. 邮政信箱 1174 WHITE PLAINS NY 10602 10409 POELLINETZ, ANDRE POELLINETZ, ANDRE PO.BOX 957332 HOFFMAN ESTATES IL 60195 6053 CARDIAC SCIENCE CORP. CARDIAC SCIENCE CORP. PO.BOX 83261 BOTHELL IL 60691-0261 5101 SPECIALTY FLOORS INC.SPECIALTY FLOORS INC. PO.BOX 8098 ROCKFORD IL 61126 15299 ROACH, ARLENA ROACH, ARLENA PO。BOX 804823 CHICAGO IL 60680 4779 CHECKMATE CHECKMATE PO。BOX 757 SAN AUGUSTINE TX 75972 5664 BENZ 显微镜光学中心 BENZ 显微镜光学中心 PO。BOX 7022 ANN ARBOR MI 48107 6533 PLATYPUS 问题 PLATYPUS 问题 PO。BOX 6957 CHAMPAIGN IL 61826 10456 BERNARDI,JIM BERNARDI,JIM PO。BOX 670 LAKE FOREST IL 60045 12618 EAGLETON SCHOOL INC。 EAGLETON SCHOOL INC。 PO。BOX 315 GREAT BARRINGTON NY 13317 6016 ILLINOIS ENVIRONMENTAL PROTECTION AGENCY ILLINOIS ENVIRONMENTAL PROTECTION AGENCY PO。BOX 19276 SPRINGFIELD IL 62794 4615 MESSMER,DALE MESSMER,DALE PO。BOX 138 CAPE FAIR MO 65624 7792 KOLLER, CHRISTINE KOLLER, CHRISTINE PO.BOX 116 PLAINFIELD IL 60544 11780 JEMHEDZ 定制服装和配饰 JEMHEDZ 定制服装和配饰 PO.订阅 ADVANTA CITRIX SYSTEMS INC.BOX 689 PLAINFIELD IL 60544 872 GORDON FLESCH GORDON FLESCH PO BOX 992 MADISON WI 53701-0992 3482 AGS PUBLICATIONS AGS PUBLICATIONS PO BOX 99 CIRCLE PINES MN 55014 5255 BLACK & DECKER BLACK & DECKER PO BOX 98692 CHICAGO IL 60693 9632 SUPER CDA SUPER CDA PO BOX 957491 HOFFMAN ESTATES IL 60195 3122 SENDRA SERVICE CORP. SENDRA SERVICE CORP. PO BOX 957 MOKENA IL 60448 3138 RAINBOW BOOK CO. RAINBOW BOOK CO. PO BOX 95432 VERNON HILLS IL 60694-5432 6094 美国学习媒体 美国学习媒体 PO BOX 95393 拉斯维加斯 NV 89193 3507 VOYAGER FLEET SYSTEMS INC。美国银行 VOYAGER FLEET SYSTEM PO BOX 952818 圣路易斯 MO 63195-2818 167 BARNES & NOBLE BARNES & NOBLE PO BOX 951610 JOLIET TX 75395-1610 745 FEDEX FEDEX PO BOX 94515 帕拉廷 IL 60094-4515 4585 ACES DEMOLITION ACES DEMOLITION PO BOX 945惠顿 IL 60189 6763 CENGAGE LEARNING CENGAGE LEARNING PO BOX 936743 亚特兰大 GA 31193-6743 519 CURRICULUM ASSOCIATES CURRICULUM ASSOCIATES PO BOX 936600 北比勒里卡 GA 31193-6600 13026 PRO CARE THERAPY, INC.PRO CARE THERAPY, INC. PO BOX 934411 亚特兰大 GA 31193-4411 7677 LEVENGER TOOLS AND SERIOUS READERS LEVENGER TOOLS AND SERIOUS READERS PO BOX 933842 亚特兰大 GA 31193 3910 CITRIX SYSTEMS INC.订阅 ADVANTA PO BOX 931686 FT. LAUDERDALE GA 31193-1686 3473 CARUS PUBLISHING CO. CARUS PUBLISHING CO. PO BOX 9306 LA SALLE IL 61301 3149 BUCKLE DOWN/OPTIONS PUBLISHING COMPANY BUCKLE DOWN/OPTIONS PUBLISHING COMPANY PO BOX 920 PO BOX 6395 NORTHBOROUGH NY 10249-6395 5316 MRA MRA PO BOX 911 PEWAUKEE WI 53072 5379 CRUTCHFIELD CRUTCHFIELD PO BOX 9032 CHARLOTTESVILLE VA 22906 7695 SOUNDBYTES SOUNDBYTES PO BOX 9022 HICKSVILLE NY 11802 1568 NASCO NASCO PO BOX 901 FORT ATKINSON WI 53538-0901 379 芝加哥论坛报 _ 芝加哥论坛报 _ PO BOX 9001157 路易斯维尔 KY 40290-1157 9280 共和国服务 共和国服务 #719 PO BOX 9001154 乔利埃特 KY 40290-1154 2389 每周读者 每周读者 PO BOX 8999。DELRAN NJ 08075-8999 3726 SHARP DECORATING SHARP DECORATING PO BOX 889 PLAINFIELD IL 60544 5857 区配送中心 区配送中心 PO BOX 888387 ATLANTA GA 30356 10035 POMP'S TIRE POMP'S TIRE PO BOX 88697 MILWAUKEE WI 53288-8697 11110 CARY COMPANY CARY COMPANY PO BOX 88670 ADDISON IL 60680-1670 13178 NOVATOO INC. NOVATOO INC. PO BOX 88478 CAROL STREAM IL 60188-8478