XiaoMi-AI文件搜索系统
World File Search Systemsequenced

DNA 作为数据存储
1. 编码。通过将独特的 DNA 核苷酸组合分配给特定的二进制位来编码信息。为了防止编码重复序列(这很难准确排序),通常使用两个或多个核苷酸来编码一个信息位。2. DNA 合成(写入数据)。DNA 由 A、T、G 和 C 核苷酸按照与编码数据相对应的序列构成。3. DNA 存储。通过封装和在稳定的温度下(从室温到 -80°C)存储,DNA 可防止降解和代码错误(通常由紫外线、湿气和氧气等环境因素引起)。4. 检索和 DNA 测序(检索和读取数据)。DNA 通常通过聚合酶链式反应 (PCR) 扩增,然后进行测序以确定核苷酸的顺序。正在开发新方法(例如“随机访问”检索),以避免在只需要部分信息时对整个 DNA 池进行测序的成本。方法可以使用“引物”选择性地靶向和扩增特定的 DNA 数据序列,或使用“条形码”序列标记 DNA 的特定部分以便更快速地检索。5. 解码。测序的 DNA 被转换或“解码”为代表原始数据的二进制代码。解码之前,可以使用纠错算法来识别和纠正在合成、保存或测序步骤中可能引入 DNA 的错误。

生态学,不仅仅是抗生素消耗,是氨基糖苷修饰酶的全球分布的主要预测指标
抽象的抗生素消耗及其滥用量在历史上并反复指出是抗生素耐药性出现和传播的主要驱动力。然而,有几个例子表明,尽管使用抗生素的使用大量降低,并且其他因素仍处于危险之中,但耐药性可能会持续存在。在这里,我们研究了氨基糖苷耐药性的时间,空间和生态分布模式,通过筛选超过160,000多个公开可用的基因组,用于编码氨基糖苷 - 修饰酶(AME基因)的27个基因簇(AME基因)。我们发现AME基因表现出非常普遍的模式:约25%的测序细菌携带AME基因。这些细菌是从所有大陆(南极)和陆地生物群落中的所有大陆进行测序,属于大量的门。通过关注1997年至2018年之间的欧洲国家,我们表明,氨基糖苷的消费对携带AME-Gene的细菌的流行率几乎没有影响,而在生物群体中观察到大多数患病率的变化。我们进一步分析了跨生物群落的抵抗组成分的相似之处:土壤,野生动植物和人类样品似乎是了解不同生态环境之间AME基因的交流的核心。在一起,这些结果支持这样的观念,即基于减少抗生素使用的介入策略应通过对交换的更强大的控制,尤其是生态系统之间的更强控制。

Illumina数据分析指南
Illumina在Imlumina平台上测序在Illumina平台上测序的cleanplex amplicon库工作流程设置建议建议使用Illumina的本地运行经理(LRM)DNA Amplicon分析模块进行分析。LRM DNA扩增子分析模块v2.1.0可在ISEQ(控制软件V1或V2),Miseq(MCS V3),NextSeq 500/550(NCS 4.0)和NextSeq 500DX(NCS 4.0)上获得。LRM DNA扩增子分析模块v3.0.0.14可在Miseq(MCS v4.0)上获得。这些说明是一个简单的摘要,描绘了如何为已经完成的运行设置分析,或设置新运行以包括分析。样品必须通过基因组进行分析以进行分析:如果在一个测序运行中存在不同的基因组,则只能设置新运行以用一个基因组进行运行分析。但是,这些样品可以在同一运行中进行测序,然后通过基因组分批分析。如果测序运行中的所有样本均来自同一基因组,则用户可以使用任何一种分析方法。此模块对齐扩增子读取针对清单文件中指定的参考,并要求变体针对目标区域。工作流程还产生了运行质量和覆盖信息的摘要报告。有用的提示和资源:●有关其他详细信息和故障排除,请参阅Illumina的最新本地运行经理DNA

S12915_020_00927_9.pdf
背景:鞭毛藻是水生生物的人,在全球海洋中特别广泛。有些人负责有毒的花朵,而另一些人生活在共生关系中,既可以作为珊瑚中的共生式共生体,要么是感染其他生物和动物的寄生虫。鞭毛菌具有非典型的大基因组(〜3至250 GB),其基因组织和基因表达模式与密切相关的Apicomplexan寄生虫截然不同。在这里,我们测序并分析了两种早期差异和同时发生的寄生虫鞭毛蛋白变形虫菌株的基因组,以阐明这种非典型基因组特征,鞭毛藻酸酯的进化和宿主专业化的出现。结果:我们使用Illumina配对的短读和牛津纳米孔技术(ONT)的长读测序方法的组合,对两种变形虫菌株(A25和A120)进行了测序,组装和注释的高质量基因组(A25和A120)。我们发现了少数可转座元素,以及短的内含子和基因间区域以及有限的基因家族,共同促进了大变形虫基因组的紧凑性,这一特征可能与寄生虫有关。大多数变形虫蛋白(A25的63.7%和A120的59.3%)没有功能分配,但我们发现许多与Dinophyceae共享的直系同源物。我们的分析表明,尽管种间蛋白质序列相似性低,但两种基因组之间的单向簇编码和高水平的同步保护的基因趋势很强。

从头读数,使用长读和简短的抛光
您将组装来自锯齿状铜菌菌株Cav1492的分离株。该菌株具有一个染色体和五个质粒。测序数据包含7,038个小小的读取,平均读取长度超过12,000 bp,一组Illumina读取了从同一菌株进行测序的读取。Illumina读取已被删除,以降低本教程中的分析时间。数据集中包含的参考基因组是由深层覆盖的PACBIO和配对末端测序数据制成的。可从https://ncbi.nlm.nih.gov/datasets/ genome/gca_001022215.1/。

使用纳米宾HMW DNA提取,PACBIO为来自多样性的高质量基因组提供了完整的端到端工作流程
摘要PACBIO测序技术提供了最完整,最准确,连续的基因组,并已被用作许多生物多样性,保护和农业类似学计划中的核心技术。在这里,我们在工作流程中提出了重大的进步,这些进步通过提供DNA隔离的方法进一步促进测序工作,并为库准备过程提供了增强的尺寸选择。这些改进应用于各种植物,昆虫和动物样品,并在新的Revio系统上进行了测序,从每个库中产生了90多个GB的数据。
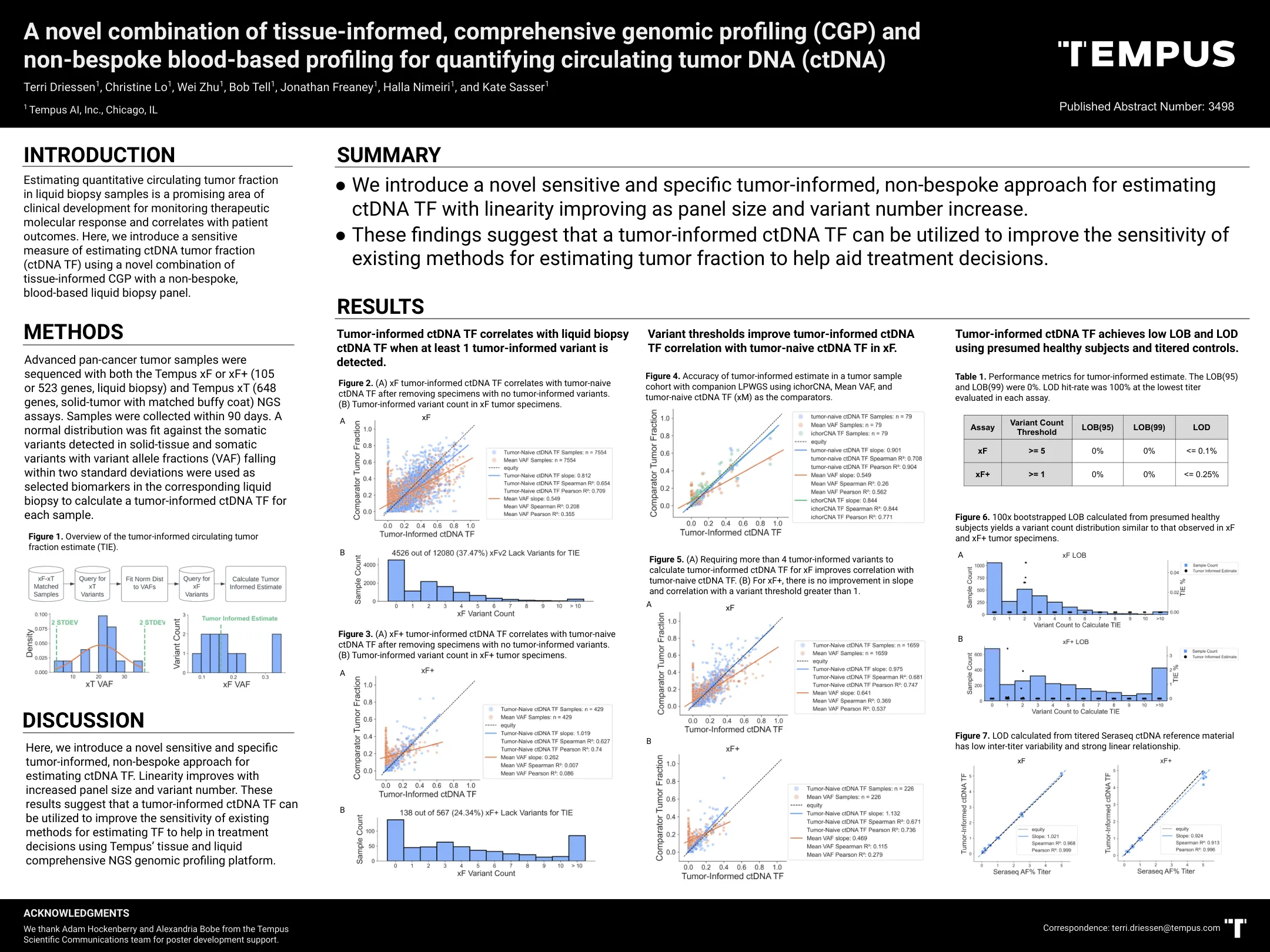
估计液体活检样品中定量循环肿瘤分数是监测治疗摩尔的临床发展领域
与TEMPUS XF或XF+(105或523基因,液体活检)和Tempus XT(648个基因,具有匹配的Buffy Coat匹配的固体肿瘤)NGS NGS测定法对晚期泛体肿瘤样品进行测序。在90天内收集样品。在固体组织和体细胞变体中检测到的躯体变异符合正态分布,并将落入两个标准偏差内的变异等位基因级分(VAF)作为相应液体活检中的选定生物标志物,以计算每个样品的肿瘤 - 信息CTDNA TF。

TotalSeq™-D1250 抗人 CD227 (MUC-1) 抗体
仅供研究使用。不可用于诊断或治疗。本产品受条款和条件(包括有限许可,位于 www.biolegend.com/terms )(“条款”)的约束,并且只能按照条款中的规定使用。在不限制上述条款的情况下,未经 BioLegend 明确书面批准,不得将 BioLegend 产品用于条款中定义的任何商业用途、以任何形式转售、用于制造、逆向工程、测序或以其他方式研究或用于了解其设计或成分。无论本文档中提供的信息如何,用户均应自行负责确定任何许可要求


开发了纳米孔测序策略,用于检测突变,在金黄色葡萄球菌菌株中赋予万古霉素中间耐药性
摘要金黄色葡萄球菌是菌血症和其他医院感染的主要原因。细胞壁活性抗生素万古霉素通常用于治疗耐甲氧西林(MRSA)和敏感(MSSA)感染。万古霉素中间的金黄色葡萄球菌(Visa)变体可以通过从头突变产生。在这里,我们进行了试点实验,以开发一种基于PCR/长阅读测序的合并的方法,用于检测先前已知的签证突变。引物旨在生成10个含量涵盖与签证表型相关的16个基因。我们对牛津纳米孔衔接子的读数长期读取,我们对据和and go骨流通量进行了测序。然后,我们通过映射读取读取的父母共识或已知参考序列,并比较称为变体与实验室选择中已知签证突变的数据库进行了比较。池中的每个扩增子被测序为高(。1,000)覆盖范围,并且在扩增子长度和覆盖范围之间未发现任何关系。我们还能够检测到因果突变(步行646c。g)在源自USA300菌株的签证突变体中(来自父母菌株N384的N384-3)。将突变体(N384-3)和父母(N384)DNA从0到1个突变体以不同的比例(N384)DNA表明平均次要等位基因频率(6.5%)的突变检测阈值在95%侧置(两个标准的差异高于平均突变频率高于平均值的频率))。该研究奠定了直接的金黄色葡萄球菌抗生素抗生素基因型基因型推断,并使用临床样品的快速纳米孔测序。