XiaoMi-AI文件搜索系统
World File Search Systemsingle

单一审计要求政策
奖项与财务报表的关系(如果有财务报表)。如果审计师根据美国注册会计师协会(AICPA)或上市公司会计监督委员会(PCAOB)的审计标准对受助者进行财务报表审计,审计师将根据 AICPA 或 PCAOB 标准(如适用)报告联邦奖项与整个财务报表的关系。4. 了解联邦奖项的内部控制结构;5. 审计并提供遵守要求的意见。当营利性获奖者获得多项联邦奖项,并且其中一项或多项奖项在一个财政年度的联邦资金支出为 750,000 美元或更多时,每个支出 750,000 美元或以上的奖项都需要进行合规审计。其余奖项不需要单独或汇总进行合规审计。总支出为 750,000 美元或以上,但未获得任何单项支出为 750,000 美元或以上的奖项的受奖人,需要对总计奖项(即作为奖项集群)进行合规审计。


超导纳米线的单片整合...
摘要我们演示了超导的单光子检测器(SPD),这些检测器(SPD)在每个像素上本地集成了信号。通过超导纳米线SPD与Josephson Electronics的单片整合来实现此能力。动机是实现具有类似于CMOS传感器对应物的集成功能的超导传感器元素。像素可以以多种模式运行。首先,我们证明可以单独计数光子,每个检测事件添加了相同数量的超频率与集成元素。第二,我们演示了一个活动增益控制选项,其中可以动态调整每个检测事件的信号以说明可变光条件。此外,像素可以无限期地保留信号以记录在集成周期内发生的所有计数,或者像素可以在衰减时间常数中记录检测事件的褪色信号。我们描述了其他半导体读数电路,该电路将在以后的工作中使用,以实现与CMOS阵列读取体系结构兼容的超导SPD的可扩展的大型传感器阵列。

呼吸机引起的单纤维细胞结构...
呼吸机诱导的隔膜功能障碍(VIDD)是需要机械通气(MV)和神经肌肉阻滞(NMBA)的重症监护单元(ICU)治疗的常见续集。它的特征是隔膜无力,延长的呼吸器断奶和不良后果。解离性糖皮质激素(例如Vamorolone,VBP-15)和伴侣共同诱导剂(例如BGP-15)先前在ICU-RAT模型中显示出积极影响。在肢体肌肉疾病肌病中,优先肌球蛋白损失占上风,而肌纤维蛋白翻译后修饰在VIDD中更为主导。尚不清楚特定力的明显下降(归一化为横截面区域)是否是收缩性信号变化的纯粹结果,或者隔膜弱点是否也通过肌球的细胞体系结构来迅速发展,以及vbp-15或BGP-15或BGP-15的范围,通过肌发光的细胞体系结构来实现结构性相关。为了解决这些问题,我们进行了无标签的多光子第二次谐波产生(SHG)成像,然后在单个diaphragm肌肉肌中进行定量形态计量学,从健康大鼠进行MV + NMBA的五天或10天的健康大鼠,以模拟ICU治疗而无需混淆病理(例如Sepsis)。大鼠每天接受泼尼松龙,VBP-15,BGP-15或无治疗。肌球蛋白-II SHG信号强度,纤维直径(FD)以及肌纤维角平行性的参数

2025-单分子生物物理
本次会议将是在阿斯彭物理中心(ACP)举行的有关单分子生物物理学(SMB)的第12个双年展研讨会,该研讨会是在2001年成功的系列上建立的。SMB会议重点介绍了单分子生物物理学领域的最新进展,包括其实验和理论前沿。主题每年有所不同。过去的会议中涵盖的生物系统包括基于核酸的酶(聚合酶,拓扑异构酶,解旋酶等。),核酸(DNA,RNA),机械酶(肌球蛋白,动力蛋白,动力蛋白,ATP合酶,鞭毛运动)以及分子生理学(折叠/展开,结合,信号传导和其他生物结构变化)的方面。精选的实验技术包括高级荧光,光学镊子,磁性镊子,扫描的探针技术,纳米孔,冷冻电子显微镜和超分辨率技术。这个研讨会传统上吸引了实验者,计算科学家和理论家的混合。

JSL可持续性报告单
气候变化的日益增长的影响在全球气温上升,极端天气事件以及对生态系统的中断,威胁着全球数百万的生计,健康和安全性。这些挑战强调了对政府,企业,公民机构,学术机构和其他人的迫切需求,以共同设计和采用手段,以减少,管理并最终阻止对环境和地球的持续破坏。最近在阿塞拜疆巴库举行的2024年联合国气候变化会议(COP29)促进了限制排放以维护巴黎协议1.5°C目标的紧迫性。cop29参与者1强调有必要增加可再生能源的采用率,到2030年全球平均年平均能量效率率两倍,并加速采用零和低排放技术,包括低碳氢,包括低碳氢,这将在脱氧而脱氧的工业型工业型脱碳中起着至关重要的作用。

检测硅中的单W-中心
控制硅中各个荧光缺陷的量子特性是对高级量子光子设备容易伸缩的关键挑战。迄今为止,研究的重点是基于硅晶格中纳入的杂质的外部缺陷。在这里,我们证明了硅中单个固有缺陷的检测,该缺陷与称为W-Center的三个互化络合物相关,在1.218 µm处的零孔子线。研究其单光子发射特性揭示了有关该常见辐射损伤中心的新信息,例如其偶极取向及其光物理学。我们还确定了其显微镜结构,并表明,尽管此缺陷不具有带隙中的电子状态,但库仑相互作用导致硅带隙下方的激子辐射重组。这些结果可能会基于硅的固有发光缺陷(例如综合量子光子学和量子通信)的固有发光缺陷为多种量子观点树立阶段。
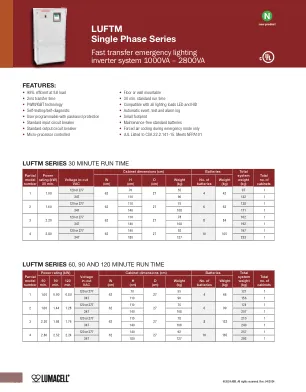
luftm单相系列
橱柜单个独立式或壁挂式NEMA 1型钢柜粉末涂有腐蚀和刮擦性。前访问设计。顶部和左侧导管带有淘汰赛。使用IGBT/PWM技术的逆变器逆变器将电池提供的直流电压转换为精确稳定振幅和频率的交流电压,适用于大多数复杂的电气设备。真正的正弦输出波形,失真非常低(线性载荷小于3%)。16个线周期的超负荷能力高达150%。充电器全自动,温度补偿,微处理器控制的充电器充电器在标称AC输入电压下最多24小时充分放电电池。交流输入电流限制和过电压保护。电池系统提供10年,无维护的密封阀调节的铅钙电池。30分钟。在正常工作温度下全负载的标准排放时间。包括低压断开保护。无需特殊通风。自我诊断自动自动测试由每月5分钟和全运行时间的年度功能组成。正式的控制面板包括4行20个字符的OLED显示屏,以及一个控制和监视内部系统的键盘。这允许操作员在发生时轻松“观察”系统功能并检查