XiaoMi-AI文件搜索系统
World File Search SystemtRNA

从犬分离出的酸性酸CACC的完整基因组序列
从犬粪便中分离出摘要酸性CACC 537,并据报道具有生物性特性。我们旨在通过功能基因组分析来表征该菌株的潜在益生菌特性。使用PACBIO RSII和Illumina平台进行了酸性P.酸性CACC 537的完整基因组测序,并包含一个带有42%G + C含量的圆形Chromo(2.0 MB)。序列是注释,显示1,897个蛋白质编码序列,15个rRNA和56个trNA。确定酸性P.酸性CACC 537基因组携带已知与免疫系统有关的基因,防御机制,限制性修饰(R-M)和CRISPR系统。CACC 537被证明有益于在发酵过程中预防病原体感染,有助于宿主免疫和维持肠道健康。这些结果可在酸性假单胞菌的地位和工业益生菌饲料添加剂的发展下进行全面,从而有助于改善宿主的免疫力和肠道健康。关键字:酸性花生,犬,全基因组测序

phi29 样 Microbacterium foliorum podovirus 噬菌体的完整基因组序列 PineapplePizza
使用氯化锌沉淀法(7)从高滴度裂解物中提取 DNA,使用 NEBNext Ultra II 试剂盒(New England Biolabs,马萨诸塞州伊普斯威奇)制备用于测序,并使用匹兹堡噬菌体研究所(宾夕法尼亚州匹兹堡)的 Illumina MiSeq 仪器(v3 试剂)进行测序。对总共 1,056,847 个单端 150 bp 读数进行 9,214 倍覆盖度的测序。分别使用 Newbler v2.9 和 Consed v29.0 进行组装和质量控制检查(8, 9)。PineapplePizza 的基因组有 16,662 个碱基对,G + C 含量为 53.6%。没有测序读数超出基因组末端,基因组末端的 101 bp 反向重复与共价结合的末端蛋白一致,如 phi29(10)。使用 NCBI BLASTn (11) 进行全基因组比对,结果显示与其他 Microbacterium 噬菌体无显著的核苷酸序列相似性,PineapplePizza 被归类为单一基因。使用 Glimmer v3.02 (12) 和 GeneMark v2.5 (13) 自动注释 PineapplePizza 的基因组,然后使用 Phamerator (14)、DNA Master v5.23.6 ( http://phagesdb.org/DNAMaster/ )、PECAAN、BLAST (11) 和 HHPred (15) 手动细化。Aragorn v1.2.38 (16) 或 tRNAscan-SE v2.0 (17) 未鉴定出 tRNA 基因。所有分析均使用默认设置进行。

Cerastium alpinum、C. arcticum 和 C. nigrescens 的完整叶绿体基因组:基因组结构、比较和系统发育分析
角菜属(Cerastium alpinum)约有 200 个物种,主要分布在北半球的温带气候中。我们在此报告了角菜(Cerastium alpinum)、北极角菜(C. arcticum)和黑色角菜(C. nigrescens)的完整叶绿体基因组。cp 基因组长度范围为 147,940 至 148,722 bp。它们的四部分环状结构具有相同的基因组织和内容,包含 79 个蛋白质编码基因、30 个 tRNA 基因和 4 个 rRNA 基因。每个物种的重复序列从 16 到 23 个不等,回文重复最为常见。每个物种已鉴定的 SSR 数量范围为 20 到 23 个,它们主要由含有 A/T 单元的单核苷酸重复组成。根据 Ka/Ks 比率值,大多数基因受到纯化选择。新测序的叶绿体基因组具有高频率的 RNA 编辑特征,包括 C 到 U 和 U 到 C 的转换。基于 71 个蛋白质编码基因的序列,重建了 Cerastium 属和石竹科内的系统发育关系。系统发育树的拓扑结构与所研究物种的系统位置一致。Cerastium 属的所有代表都聚集在一个分支中,而 C. glomeratum 与其他分支的相似性最小。

通过对宿主来源的转移 RNA 片段进行化学修饰来靶向核梭杆菌
宿主粘膜屏障拥有一系列防御分子,以维持宿主-微生物体内平衡,例如抗菌肽和免疫球蛋白。除了这些已证实的防御分子外,我们最近还报道了人类口腔角质形成细胞与具核梭杆菌 (Fn) 之间的小 RNA (sRNA) 介导的相互作用,Fn 是一种口腔致病菌,在口腔外疾病中的影响越来越大。具体而言,在 Fn 感染后,口腔角质形成细胞会释放 Fn 靶向 tRNA 衍生的 sRNA (tsRNA),这是一类具有基因调控功能的新兴非编码 sRNA。为了探索 tsRNA 的潜在抗菌活性,我们对 Fn 靶向 tsRNA 的核苷酸进行了化学修饰,并证明所得的 tsRNA 衍生物(称为 MOD-tsRNA)在纳摩尔浓度范围内无需任何运载工具即可对各种 Fn 型菌株和临床肿瘤分离株表现出生长抑制作用。相反,相同的 MOD-tsRNA 不会抑制其他代表性口腔细菌。进一步的机制研究揭示了 MOD-tsRNA 在抑制 Fn 中的核糖体靶向功能。总之,我们的工作提供了一种通过共同选择宿主衍生的细胞外 tsRNA 来靶向致病菌的工程方法。
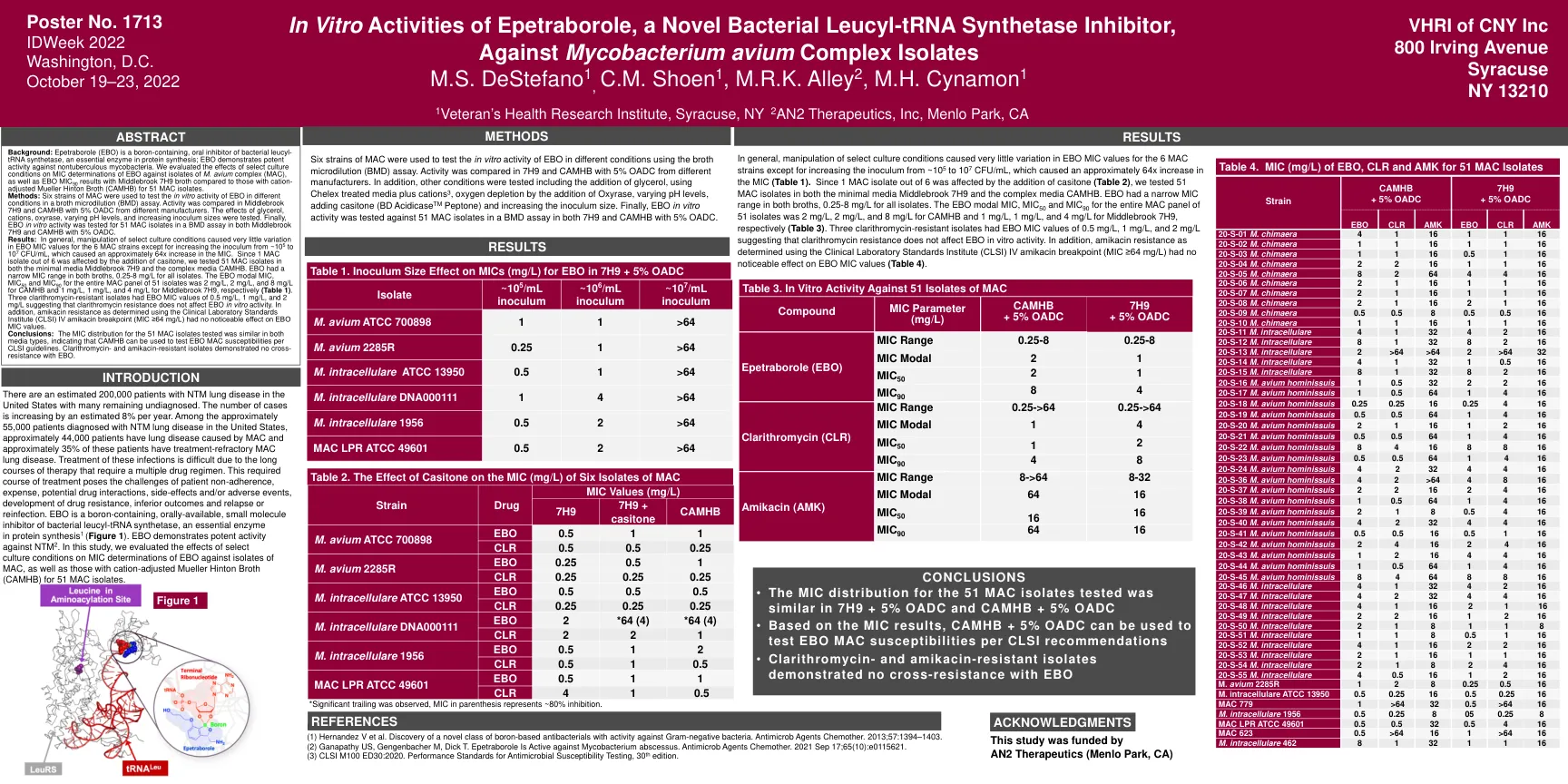
Epetraborole的体外活性,一种新型细菌亮氨基...
美国估计有200,000例NTM肺部疾病患者,许多人仍未诊断。案件数量估计每年增加8%。在美国诊断出患有NTM肺部疾病的大约55,000例患者中,大约44,000名患者患有MAC引起的肺部疾病,其中约35%患有治疗难治性的MAC肺疾病。由于需要多种药物方案的长期治疗,因此很难治疗这些感染。这种必需的治疗过程构成了患者不遵守,费用,潜在的药物相互作用,副作用和/或不良事件的挑战,耐药性的发展,劣质结果以及复发或再感染。eBO是一种含硼的,口服可用的小分子抑制剂的细菌亮氨基-TRNA合成酶,这是蛋白质合成1中必不可少的酶(图1)。EBO证明了针对NTM 2的有效活性。在这项研究中,我们评估了精选培养条件对EBO对MAC分离株的MIC测定的影响,以及阳离子调整后的Mueller Hinton Broth(CAMHB)对51个MAC分离株的影响。

rnagrail:RNA 3D结构预测的图形神经网络和扩散模型
RNA的功能与X射线晶体学,NMR和Cryo-EM传统上探索的3D结构本质上息息相关。但是,这些实验通常缺乏原子水平的分辨率,从而使需要准确的RNA RNA结构预测工具。这一需求推动了人工智能(AI)的进步,该技术已经彻底改变了蛋白质结构的预测。不幸的是,由于稀疏和不平衡的结构数据,RNA场中的类似突破仍然有限。在这里,我们介绍了RNAGRAIL,这是一种新型的RNA 3D结构预测方法,该方法侧重于使用denoising扩散概率模型(DDPM)进行RNA子结构。与Alphafold 3(AF3)不同,被许多人认为是Oracle,Rnagrail允许专家用户定义基本对约束,从而提供出色的灵活性和精确度。,我们的方法在平均RMSD方面优于AF3,而平均ERMSD的表现为24%。此外,就相互作用网络保真度(INF)而言,它完美地再现了规范的二级结构优于AF3。rnagrail表现出各种RNA图案和家庭的鲁棒性。尽管受过rRNA和tRNA的训练,但它有效地概括为新的RNA家族,因此解决了RNA 3D结构预测中的主要挑战之一。这些结果强调了专注于小的RNA组件并集成用户定义的约束以显着增强RNA 3D结构预测的潜力,从而在RNA建模中设定了新标准。

建模蛋白质折叠机可以如何工作
蛋白质在体内稳健且可重复地折叠,但许多人不能在体外与细胞成分分离折叠。在体外或体内,蛋白质本地构象的途径仍然很大未知。硅质中概括蛋白质折叠途径的缓慢进展可能表明我们对折叠的理解在自然界中的理解中的基本缺陷。在这里,我们认为活细胞中的蛋白质折叠可能仅由Gibbs自由能的减少驱动,并提出应将蛋白质折叠在体内建模为活性能量依赖性过程。这种蛋白质折叠机的作用机理可能包括直接操纵肽主链。为了显示蛋白质折叠机的可行性,我们进行了分子动力学模拟,通过使用机械力来旋转C-末端氨基酸,同时限制了N末端氨基酸运动,从而增强了分子动力学模拟。值得注意的是,将这种简单的肽骨架对标准分子动力学模拟的简单操纵的引入确实促进了五种不同α-螺旋肽的天然结构的形成。这种效果可能在体内的共同翻译蛋白折叠期间起作用:考虑到核糖体的肽基转移酶中心tRNA 3'-end的旋转运动,这种运动可能会引入新的肽,并以类似于我们的模拟方式以类似的方式影响肽的折叠路径。

多重引导RNA表达系统及其对植物基因组工程的功效
摘要背景:化脓性链球菌 CRISPR 系统由 Cas9 内切酶 (Sp Cas9) 和含有靶标特异性序列的单链向导 RNA (gRNA) 组成。理论上,Sp Cas9 蛋白可以切割与基因组中结合的 gRNA 一样多的靶标位点。结果:我们引入了一种无 PCR 的多 gRNA 克隆系统来编辑植物基因组。该方法包括两个步骤:(1)在 pGRNA 载体中的 tRNA 和 gRNA 支架序列之间克隆两个单链寡核苷酸片段的退火产物,该片段的每条链上都含有互补的靶标结合序列;(2)使用 Golden Gate 组装方法将来自几个 pGRNA 载体的 tRNA-gRNA 单元与含有 Sp Cas9 表达盒的植物二元载体组装在一起。我们通过进行靶向深度测序验证了多重 gRNA 表达系统在野生烟草(Nicotiana attenuata)原生质体和转化植物中的编辑效率和模式。Sp Cas9-gRNA 的两次近端切割大大提高了编辑效率,并在两个切割位点之间诱导了较大的缺失。结论:这种多重 gRNA 表达系统能够高通量生产单个二元载体,并提高植物基因组编辑的效率。关键词:CRISPR-Cas9、金门组装、多重 gRNA、植物基因组编辑

来自哈萨克斯坦的七种假发物种的叶绿体基因组的比较分析
杜松种类是杯形科中的灌木或树木,在森林生态系统中起着重要作用。在这项研究中,我们报告了在哈萨克斯坦收集的五种假发物种的质体(PT)基因组的完整序列(j。 communis,j。 Sibirica,J。 pseudosabina,j。 semiglobosa和j。 Davurica)。 除了两个完整的Pt基因组序列外,还注释了五种物种的Pt基因组的序列。 Sabina和J。 Seravschanica,我们先前已报告。 将这七种物种的Pt基因组序列与Pub-lic ncbi数据库中可用的杜松物种的Pt基因组进行了比较。 杜松物种的PT基因组的总长度,包括先前发表的PT基因组数据,范围为127,469 bp(j。 semiglobosa)至128,097 bp(j。 communis)。 每个杜松子质体由119个基因组成,包括82个蛋白质编码基因,33个转移RNA和4个核糖体RNA基因。 在确定的基因中,16个包含一个或两个内含子,并复制了2个tRNA基因。 对PT基因组序列的比较评估表明,鉴定了1145个简单序列重复标记。 基于82种蛋白质编码基因的26种假发物种的系统发育树,将杜松样品分为两个主要进化枝,对应于Juniperus和Sabina切片。 PT基因组序列的分析表明ACCD和YCF2是两个最多态性基因。在这项研究中,我们报告了在哈萨克斯坦收集的五种假发物种的质体(PT)基因组的完整序列(j。communis,j。Sibirica,J。 pseudosabina,j。 semiglobosa和j。 Davurica)。 除了两个完整的Pt基因组序列外,还注释了五种物种的Pt基因组的序列。 Sabina和J。 Seravschanica,我们先前已报告。 将这七种物种的Pt基因组序列与Pub-lic ncbi数据库中可用的杜松物种的Pt基因组进行了比较。 杜松物种的PT基因组的总长度,包括先前发表的PT基因组数据,范围为127,469 bp(j。 semiglobosa)至128,097 bp(j。 communis)。 每个杜松子质体由119个基因组成,包括82个蛋白质编码基因,33个转移RNA和4个核糖体RNA基因。 在确定的基因中,16个包含一个或两个内含子,并复制了2个tRNA基因。 对PT基因组序列的比较评估表明,鉴定了1145个简单序列重复标记。 基于82种蛋白质编码基因的26种假发物种的系统发育树,将杜松样品分为两个主要进化枝,对应于Juniperus和Sabina切片。 PT基因组序列的分析表明ACCD和YCF2是两个最多态性基因。Sibirica,J。pseudosabina,j。semiglobosa和j。Davurica)。 除了两个完整的Pt基因组序列外,还注释了五种物种的Pt基因组的序列。 Sabina和J。 Seravschanica,我们先前已报告。 将这七种物种的Pt基因组序列与Pub-lic ncbi数据库中可用的杜松物种的Pt基因组进行了比较。 杜松物种的PT基因组的总长度,包括先前发表的PT基因组数据,范围为127,469 bp(j。 semiglobosa)至128,097 bp(j。 communis)。 每个杜松子质体由119个基因组成,包括82个蛋白质编码基因,33个转移RNA和4个核糖体RNA基因。 在确定的基因中,16个包含一个或两个内含子,并复制了2个tRNA基因。 对PT基因组序列的比较评估表明,鉴定了1145个简单序列重复标记。 基于82种蛋白质编码基因的26种假发物种的系统发育树,将杜松样品分为两个主要进化枝,对应于Juniperus和Sabina切片。 PT基因组序列的分析表明ACCD和YCF2是两个最多态性基因。Davurica)。除了两个完整的Pt基因组序列外,还注释了五种物种的Pt基因组的序列。Sabina和J。Seravschanica,我们先前已报告。将这七种物种的Pt基因组序列与Pub-lic ncbi数据库中可用的杜松物种的Pt基因组进行了比较。杜松物种的PT基因组的总长度,包括先前发表的PT基因组数据,范围为127,469 bp(j。semiglobosa)至128,097 bp(j。communis)。每个杜松子质体由119个基因组成,包括82个蛋白质编码基因,33个转移RNA和4个核糖体RNA基因。在确定的基因中,16个包含一个或两个内含子,并复制了2个tRNA基因。对PT基因组序列的比较评估表明,鉴定了1145个简单序列重复标记。基于82种蛋白质编码基因的26种假发物种的系统发育树,将杜松样品分为两个主要进化枝,对应于Juniperus和Sabina切片。PT基因组序列的分析表明ACCD和YCF2是两个最多态性基因。使用这两个基因对26种假发物种的系统发育评估证实,它们可以有效地用作该属中植物分析的DNA条形码。这些假发物种的测序质体提供了大量遗传数据,这些数据对于该属的将来的基因组研究很有价值。

这是以下文章的同行评审版本:Ranzau、Brodie L.、Rallapalli、Kartik L.、Evanoff、Mallory、Paesani、Francesco、Komor、Ale
碱基编辑器是一种基因组编辑工具,可通过对 DNA 中的核碱基进行化学修饰来实现位点特异性碱基转换。腺嘌呤碱基编辑器 (ABE) 利用腺苷脱氨酶将目标腺苷修饰为肌苷中间体,从而将 DNA 中的 A•T 转换为 G•C 碱基对。由于缺乏可以修饰 DNA 的天然腺苷脱氨酶,ABE 是从 tRNA 脱氨酶 TadA 进化而来的。之前利用由野生型 (wt) TadA 组成的 ABE 进行的实验未显示对 DNA 的可检测活性,因此需要定向进化以使该酶能够接受 DNA 作为底物。在这里,我们表明 wtTadA 可以在细菌和哺乳动物细胞中的 DNA 中进行碱基编辑,对 TAC 的序列基序有严格的要求。我们利用这一发现优化了报告基因检测,以检测低至 0.01% 的碱基编辑水平。最后,我们将该分析与完整 ABE:DNA 复合物的分子动力学模拟结合使用,以更好地了解突变 TadA 变体的序列识别如何随着它们积累突变而变化,从而更好地编辑 DNA 底物。