XiaoMi-AI文件搜索系统
World File Search Systemvariation




变更通知 Dogger Bank Teesside AB OWF 附表 8 变更 3
2021 年 1 月 28 日,海洋管理组织 (MMO) 收到苏格兰电力可再生能源公司的请求,要求更改《2017 年东英吉利三处海上风电场命令》(“命令”)附表 15 中的“视同海洋许可证”(“DML”)。该请求旨在重新分配东英吉利三处 DML 中的同意处置许可。

变更通知 Dogger Bank Teesside AB OWF SChedule 8 变更 3
2021 年 1 月 28 日,海洋管理组织 (MMO) 收到苏格兰电力可再生能源公司的请求,要求更改 2017 年东英吉利三处海上风电场命令 (“命令”) 附表 14 中包含的视同海洋许可证 (DML)。该请求旨在重新分配 EA 三处 DML 内的同意处置许可。

变更通知 Dogger Bank Teesside AB OWF 附表 8 变更 3
如果拟使用打入或部分打入桩基础,则在向 MMO 提交符合《东英吉利三号项目南北海 pSAC 场地完整性计划》中原则上所列原则的《东英吉利三号项目南北海 pSAC 场地完整性计划》且 MMO 确信该计划提供了必要的缓解措施以避免对相关场地的完整性(根据 2007 年法规的含义)造成不利影响(只要港湾鼠海豚是该场地的受保护特征)之前,不得开始许可活动或这些活动的任何阶段。
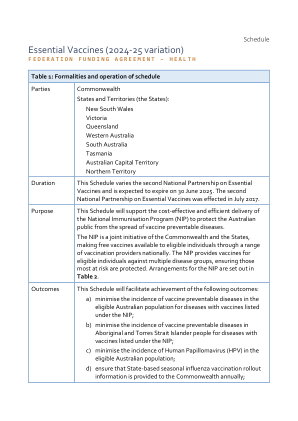
基本疫苗(2024-25 年变化)
• 联邦自用费用包括采购 NIP 涵盖的所有疫苗。本附表不包括 COVID-19 疫苗。 • 根据本协议条款,各州无需提供资金或实物捐助。但是,由于各州负责实现本协议下的成果,因此各州分配自己的资金来源并相应地提供实物捐助,包括支持本协议资助的服务和活动。 • 估计的国家伙伴关系支付数字基于 2023-24 财年的联邦自用支出金额(6.414 亿美元)。