XiaoMi-AI文件搜索系统
World File Search System亲和力



基于柔软且灵活材料的亲和力传感器
本期刊文章的自存档后印本版本可在林雪平大学机构知识库 (DiVA) 上找到:http://urn.kb.se/resolve?urn=urn:nbn:se:liu:diva-169862 注意:引用本研究时,请引用原始出版物。Meng, L., Turner, AP, Mak, WC, (2020), 基于软性和柔性材料的亲和力传感器, Biotechnology Advances, 39, 107398. https://doi.org/10.1016/j.biotechadv.2019.05.004

两个半胱氨酸分子的高亲和力结合......
荧光滴定表明,人类低分子量激肽原 (LK) 能以高亲和力结合两分子的蛋白酶 L 和 S 以及木瓜蛋白酶。相比之下,第二分子的蛋白酶 H 的结合要弱得多。通过滴定法(监测酶活性损失和沉降速度实验)证实了 2:1 的结合化学计量。蛋白酶 L 和 S 与木瓜蛋白酶的结合动力学表明,两个蛋白酶结合位点的结合速率常数 k,,,,, = 10.7-24.5 x 106 M" sI 和 k,,,,, = 0.83-1.4 x 106 M" s-'。将这些动力学常数与完整 LK 及其分离结构域的先前数据进行比较,表明结合较快的位点也是结合较紧的位点,位于结构域 3 上,而结合较慢、亲和力较低的位点位于结构域 2 上。这些结果还表明,两个结合位点之间或来自激肽原轻链的蛋白酶结合没有明显的空间障碍。

基于一致性评价的药物-靶标亲和力预测方法...
摘要 - 药物发现的第一步是找到具有针对特定靶标的药用活性的药物分子部分。因此,研究药物靶标蛋白与小化学分子之间的相互作用至关重要。然而,传统的发现潜在小药物分子的实验方法劳动密集且耗时。目前,人们对使用药物分子相关数据库建立计算模型来筛选小药物分子非常感兴趣。在本文中,我们提出了一种使用深度学习模型预测药物靶标结合亲和力的方法。该方法使用改进的GRU和GNN分别从药物靶标蛋白序列和药物分子图中提取特征以获得它们的特征向量。组合向量用作药物-靶标分子对的向量表示,然后输入到完全连接的网络中以预测药物-靶标结合亲和力。该提出的模型证明了其在DAVIS和KIBA数据集上预测药物-靶标结合亲和力的准确性和有效性。

4,6-二取代嘧啶基微管亲和力-...
杂环化合物在合成和天然化学空间中普遍存在,是各种应用的基本骨架(Reymond,2015)。杂环化合物意义重大,因为它们对人类、植物和动物至关重要(Katritzky 等人,2010)。在广泛的中小型杂环化合物中,嘧啶核构成了一组重要的药理活性化合物(Das 等人,2022)。该核心的重要性得到了充分的支持,因为它是核碱基(胞嘧啶、胸腺嘧啶、尿嘧啶)以及许多临床批准药物的片段。例如,嘧啶核存在于 5-氟尿嘧啶、伊马替尼(抗癌药)、利匹韦林(抗病毒药)、艾克拉普林(抗生素)、甲氧苄啶(抗菌药)和许多其他药物中(Nammalwar and Bunce,2024 年)。此外,它能够充当生物电子等排体(用于芳香核)并通过非共价相互作用 (NCI) 与生物靶标相互作用,使其成为药物发现计划的绝佳候选者(Nammalwar and Bunce,2024 年)。大量研究表明,嘧啶是开发针对慢性和传染病的药物的有希望的支架(Nadar and Khan,2022 年)。近年来,已鉴定出几种具有抗原虫(Rahman 等人,2024;Singh 等人,2024)、抗炎(Fatima 等人,2023)、抗神经炎症(Manzoor 等人,2023)和碳酸酐酶抑制(Manzoor 等人,2021a)活性的 4,6-二取代嘧啶。一个多世纪前就有报道,阿尔茨海默病 (AD) 现已成为痴呆症最普遍的原因,全球已报告数百万例病例。这导致了巨大的经济和人力负担(Bell,2023;Gustavsson 等人,2023)。到 2050 年,患有 AD 和其他痴呆症的人数估计将超过 1.52 亿(Nichols 等人,2022 年)。为了对抗这种使人衰弱的疾病,研究人员正在采用各种方法,其中一种方法是开发针对一种或多种 AD 机制(例如 β-淀粉样斑块、神经纤维缠结)的小分子(Takahashi 等人,2017 年)。在迄今为止鉴定出的不同类别的小分子中,基于嘧啶的化合物成为一种有希望的候选化合物(Singh 等人,2021 年;Das 等人,2022 年)。例如,Nain 及其同事(Pant 等人,2024 年)报道了一系列取代的


114-药物目标亲和力的定量预测模型...
关键字:ENROFLOXATIN,牛血清白蛋白,绑定相互作用1。引言属于氟喹诺酮类药物组的Enrorofloxin(EFLX)已被发现抑制包括铜绿假单胞菌和肠杆菌科在内的大多数革兰氏阴性病原体。在医学方面,它可用于呼吸道感染,淋病,细菌性胃肠炎,皮肤软组织感染,简单和复杂的尿路感染,尤其是由革兰氏阴性和革兰氏阳性引起的感染[1]。作为循环系统中最丰富的蛋白质之一,血清白蛋白是血液中的主要传输蛋白,可以可逆地与小分子结合,例如脂肪酸,氨基酸,药物和无机离子[2]。胆红素,激素和外源或内源性配体。HSA的X射线晶体学分析表明,此HSA的X射线晶体学分析表明,此
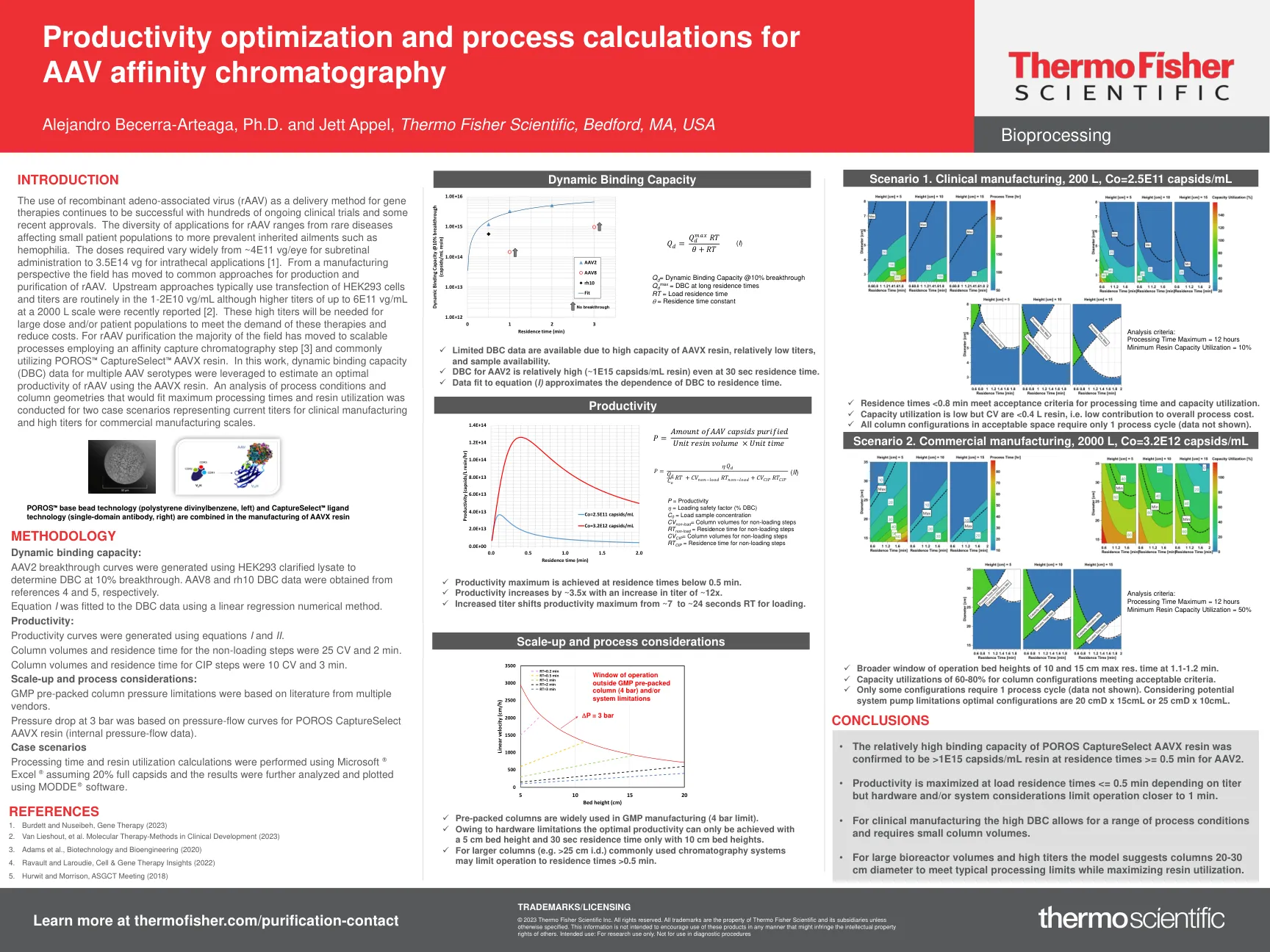
AAV亲和力色谱法的生产力优化和过程计算
使用重组腺相关病毒(RAAV)作为基因疗法的递送方法,继续使用数百项正在进行的临床试验和一些最近的批准成功。RAAV应用的多样性范围从影响小患者人群的罕见疾病到更普遍的遗传性疾病,例如血友病。剂量从〜4E11 VG/眼睛的视网膜下给药到3.5E14 VG的剂量差异很大[1]。从制造业的角度来看,该领域已转向了Raav的生产和纯化方法。上游方法通常使用HEK293细胞的转染,并且滴度通常在1-2e10 Vg/ml中使用,尽管最近报道了2000 l量表的高达6E11 VG/mL的较高滴度[2]。大剂量和/或患者种群需要这些较高的滴度来满足这些疗法的需求并降低成本。对于RAAV纯化,该领域的大多数已移至使用亲和力捕获色谱步骤[3]的可扩展过程,并通常使用Poros™Capturelect™AAVX树脂。在这项工作中,利用了多种AAV血清型的动态结合能力(DBC)数据,以估算使用AAVX树脂的最佳生产力。对符合最大处理时间和树脂利用率的过程条件和柱几何形状的分析,用于两种情况,代表了用于临床制造的当前滴度和用于商业制造量表的高滴度。

逃避抗体和ACE2亲和力之间的微妙平衡
Giandown如何,1,2,3,20, * Iste Dijocite-guraluc,4,20 Chang Liu,4,5,20 Daming Zhou,2,5 Helen M. Ginn,6 Rocksha Das,4 Pather Supersa,4 Muneestan,4 Muneestan Selever,4 rungtiwa nutail,4 achcare,4 nutg nutg nutg nutg a achane nutgti a achane tak a a achane tak ta a ach a ach a a achan n achci a anc n achci a anc n achci a anc n. 4 nutg a anc Acaria,4 4 Rungtiwa Nuter,4 A Ofhaccane,4 4 4 Turkhn,Helen M.E. 4 ),s@strbi.ox.ac.uk(e.e.f. ),renders.ax.ac。 英国(J.R ),garve.screton@medsci.ox.ac.uk(G.R.S.) https://doi.org/10.1016/j.celrep.2022.111903),s@strbi.ox.ac.uk(e.e.f.),renders.ax.ac。 英国(J.R ),garve.screton@medsci.ox.ac.uk(G.R.S.) https://doi.org/10.1016/j.celrep.2022.111903英国(J.R),garve.screton@medsci.ox.ac.uk(G.R.S.)https://doi.org/10.1016/j.celrep.2022.111903