XiaoMi-AI文件搜索系统
World File Search System列名

4 美国食品和药品 - accessdata.fda.gov
请注意,FDA 发布的实质等同性认定并不意味着 FDA 已认定您的器械符合该法案的其他要求或其他联邦机构管理的任何联邦法规。您必须遵守该法案的所有要求,包括但不限于:注册和列名(21 CFR 第 807 部分);标签(21 CFR 第 801 部分);器械的医疗器械报告(医疗器械相关不良事件报告)(21 CFR 803)或组合产品的上市后安全报告(21 CFR 4,子部分 B)(请参阅https://www.fda.gov/combination-products/guidance-regulatory-information/postmarketing-safety-reporting-combination-products);器械质量体系 (QS) 法规(21 CFR 第 820 部分)中规定的良好生产规范要求或组合产品的现行良好生产规范(21 CFR 4,子部分 A);以及如果适用的话,电子产品辐射控制规定(该法第531-542节);21 CFR 1000-1050。

日本医疗相关产品的全球拓展...
世界卫生组织 (WHO) 于 2020 年 1 月将 2019 年冠状病毒病 (COVID-19) 宣布为国际关注的突发公共卫生事件 (PHEIC) ( 1 )。为应对这一流行病,全球开始开发针对 COVID-19 的药物、疫苗和体外诊断产品。紧急使用清单 (EUL) 是 WHO 在 PHEIC 期间评估未经许可的药物、疫苗和体外诊断产品的程序,以加快向有需要的人提供这些产品 ( 2 )。EUL 是由紧急使用评估和列名机制建立和改革的,该机制是为了应对 2014-2016 年埃博拉病毒病疫情而制定的 ( 3 )。EUL 指出了紧急情况下临时使用未经许可产品的特定标准。因此,各制药公司竞相开发 COVID-19 产品,并向 EUL 提交产品文件,以便在全球范围内供应产品。然而,在提交文件之前,世卫组织、外部专家和产品生产地的国家监管机构(NRA)之间必须建立评估平台。EUL 程序基于公私合作伙伴关系。
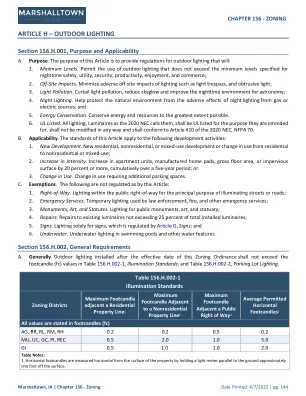
条款 H - 户外照明
A. 目的。本条款旨在为室外照明提供法规,其将:1. 最低水平。允许使用不超过为夜间安全、实用、保障、生产力、娱乐和商业而规定的最低水平的室外照明;2. 场外影响。尽量减少照明对场外造成的不利影响,例如光侵入和干扰光;3. 光污染。减少光污染,减少天辉,改善天文观测的夜间环境;4. 夜间照明。帮助保护自然环境免受燃气或电源夜间照明的不利影响;以及 5. 节能。最大程度地节约能源和资源。6. UL 列名。所有照明(2020 NEC 称之为灯具)均应通过 UL 认证,且适用于其预期用途,不得以任何方式进行修改,并应符合 2020 NEC 第 410 条、NFPA 70。B. 适用性。本条的标准适用于以下开发活动:1. 新开发项目。新的住宅、非住宅或混合用途开发项目或从住宅变为非住宅或混合用途的用途变更;2. 强度增加。在五年内累计将公寓单元、活动房屋垫块、总建筑面积或不透水表面增加 20% 或更多;或 3. 用途变更。用途变更需要增加停车位。C. 豁免。以下情况不受本条管制:1. 通行权。公共通行权内的照明,主要用于照亮街道或道路;2. 紧急服务。执法、消防和其他应急服务部门使用的临时照明; 3. 纪念碑、艺术品和雕像 。公共纪念碑、艺术品和雕像的照明; 4. 维修 。对现有灯具的维修不超过总安装灯具的 25%; 5. 标志 。仅用于标志的照明,受标志 G 条的管制;以及 6. 水下 。游泳池和其他水景中的水下照明。

K212194.pdf - accessdata.fda.gov
探针和导航活检针 法规编号:21 CFR 882.4560 法规名称:立体定位仪 监管类别:II 类 产品代码:HAW 日期:2023 年 1 月 13 日 收讫日期:2023 年 1 月 17 日 亲爱的 Bryan Hann: 我们已审查了您根据第 510(k) 条提交的上市前通知,该通知表明您有意销售上述器械,并已确定该器械与在 1976 年 5 月 28 日(医疗器械修正案颁布日期)之前在州际贸易中合法销售的同类器械或已根据《联邦食品药品和化妆品法案》(法案)的规定重新分类的器械基本等同,这些器械不需要获得上市前批准申请(PMA)批准。因此,您可以根据该法案的一般控制规定销售该器械。虽然本函将您的产品称为设备,但请注意,一些已获准的产品可能是组合产品。位于 https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm 的 510(k) 上市前通知数据库可识别组合产品提交。该法案的一般控制条款包括年度注册、设备列表、良好生产规范、标签以及禁止贴错标签和掺假的要求。请注意:CDRH 不会评估与合同责任担保相关的信息。但我们提醒您,设备标签必须真实且不得误导。如果您的设备被归类(见上文)为 II 类(特殊控制)或 III 类(PMA),则可能会受到其他控制。影响您设备的现有主要法规可在《联邦法规》第 21 篇第 800 至 898 部分中找到。此外,FDA 可能会在《联邦公报》上发布有关您设备的进一步公告。请注意,FDA 发布实质等效性判定并不意味着 FDA 已判定您的设备符合该法案的其他要求或其他联邦机构管理的任何联邦法规和规章。您必须遵守该法案的所有要求,包括但不限于:注册和列名(21 CFR 第 807 部分);标签(21 CFR 第

固德威逆变器GW15KT-DT GW15KF-DT
公司信息 环保署所有者:苏州固德威科技有限公司 联系人:蒋涛,邮箱:tao.jiang@goodwe.com 机构描述:苏州固德威科技有限公司成立于2010年11月,总部位于中国苏州高新区。公司是一家集光伏并离网逆变电源、风电并离网逆变电源、光伏控制逆变器等新能源设备研发、生产和销售为一体的高科技公司。主营业务产品包括光伏并网逆变器、光伏储能逆变器、智能数据采集器以及SEMS智能能源管理系统。固德威长期专注于太阳能、储能等新能源电源设备的研发、生产和销售。致力于为家庭、工商业用户、地面电站提供智慧能源管理等综合解决方案。公司产品通过数十项相关认证及政府列名,已大规模销售至全球100多个国家和地区,累计安装量达35GW,广泛应用于民用及商用屋顶、大型地面项目,市场强劲。公司在澳大利亚、英国、韩国、德国、荷兰、西班牙、印度、土耳其、巴西、墨西哥、美国、日本等地设立子公司或客服中心,业绩获得国际认可,在权威IHS排名中成为全球十大逆变器品牌之一。根据国际知名电力及可再生能源研究机构Wood Mackenzie统计,2018年公司在全球光伏逆变器市场排名第七,2019年公司户用储能逆变器出货量位居全球第一。产品相关或管理体系相关认证: ISO9001 ISO14001 ISO45001 ISO50001 生产场地名称及地点: 工厂一:固德威科技有限公司,地址:苏州市新区紫金路90号,邮编215011,中国 工厂二:固德威(广德)电源科技有限公司,地址:安徽省广德市通瑞东路208号 产品信息 产品名称: 逆变器GW15KT-DT(代表产品) 逆变器GW15KF-DT 产品标识:

恒运智能股份有限公司 王提浩 博士 首席技术官...
贸易/设备名称:EFAI RTSuite CT HN 分割系统 法规编号:21 CFR 892.2050 法规名称:医学图像管理和处理系统 监管类别:II 类 产品代码:QKB 日期:2022 年 1 月 28 日 收到日期:2022 年 1 月 31 日 亲爱的王 Ti-Hao: 我们已审查了您根据第 510(k) 条提交的上市前通知,该通知意在销售上述设备,并已确定该设备与在 1976 年 5 月 28 日(医疗器械修正案颁布日期)之前在州际贸易中合法销售的同类设备基本等同(就附件中规定的用途而言),或与根据《联邦食品、药品和化妆品法案》(法案)的规定重新分类的设备基本等同,这些设备不需要获得上市前批准申请(PMA)的批准。因此,您可以销售该设备,但须遵守该法案的一般控制规定。虽然本函将您的产品称为设备,但请注意,一些已获准的产品可能是组合产品。位于 https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm 的 510(k) 上市前通知数据库可识别组合产品提交。该法案的一般控制条款包括年度注册、设备列表、良好生产规范、标签以及禁止贴错标签和掺假的要求。请注意:CDRH 不会评估与合同责任担保相关的信息。但我们提醒您,设备标签必须真实且不得误导。如果您的设备被归类(见上文)为 II 类(特殊控制)或 III 类(PMA),则可能会受到其他控制。影响您设备的现有主要法规可在《联邦法规》第 21 篇第 800 至 898 部分中找到。此外,FDA 可能会在《联邦公报》上发布有关您设备的进一步公告。请注意,FDA 发布实质等效性判定并不意味着 FDA 已判定您的设备符合该法案的其他要求或其他联邦机构管理的任何联邦法规和规章。您必须遵守该法案的所有要求,包括但不限于:注册和列名(21 CFR 第 807 部分);标签(21 CFR 第

2020 年 10 月 30 日 Whiterabbit.ai Inc.先生。 Jason 其 CTO 和
贸易/设备名称:WRDensity by Whiterabbit.ai 法规编号:21 CFR 892.2050 法规名称:图片存档和通信系统 监管类别:II 类 产品代码:QIH 日期:2020 年 9 月 29 日 收讫日期:2020 年 9 月 30 日 亲爱的苏先生: 我们已审查了您根据第 510(k) 条提交的上市前通知,该通知表明您有意销售上述设备,并已确定该设备与 1976 年 5 月 28 日(医疗器械修正案颁布日期)之前在州际贸易中合法销售的同类设备基本等同(就附件中注明的用途而言),或与根据《联邦食品药品和化妆品法案》(法案)的规定重新分类的设备基本等同,这些设备不需要获得上市前批准申请(PMA)的批准。因此,您可以营销该设备,但须遵守该法案的一般控制规定。虽然本函将您的产品称为设备,但请注意,一些已获准的产品可能是组合产品。位于 https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm 的 510(k) 上市前通知数据库可识别组合产品提交。该法案的一般控制条款包括年度注册、设备列表、良好生产规范、标签以及禁止贴错标签和掺假的要求。请注意:CDRH 不会评估与合同责任担保相关的信息。但我们提醒您,设备标签必须真实且不得误导。如果您的设备被归类(见上文)为 II 类(特殊控制)或 III 类(PMA),则可能会受到其他控制。影响您设备的现有主要法规可在《联邦法规》第 21 篇第 800 至 898 部分中找到。此外,FDA 可能会在《联邦公报》上发布有关您设备的进一步公告。请注意,FDA 发布实质等效性判定并不意味着 FDA 已判定您的设备符合该法案的其他要求或其他联邦机构管理的任何联邦法规和规章。您必须遵守该法案的所有要求,包括但不限于:注册和列名(21 CFR 第 807 部分);标签(21 CFR 第

Qure.ai Technologies 2021 年 7 月 30 日 Pooja Rao 主管 ...
贸易/设备名称:qER-Quant 法规编号:21 CFR 892.2050 法规名称:医学图像管理和处理系统 监管类别:II 类 产品代码:QIH 日期:2021 年 6 月 30 日 收到日期:2021 年 7 月 1 日 亲爱的 Pooja Rao: 我们已审查了您根据第 510(k) 条提交的上市前通知,该通知表明您有意销售上述设备,并已确定该设备与在 1976 年 5 月 28 日(医疗器械修正案颁布日期)之前在州际贸易中合法销售的同类设备基本等同(就附件中规定的使用指征而言),或与根据《联邦食品、药品和化妆品法案》(法案)的规定重新分类的设备基本等同,这些设备不需要获得上市前批准申请 (PMA) 的批准。因此,您可以根据该法案的一般控制规定销售该设备。虽然这封信将您的产品称为设备,但请注意,一些已获准的产品可能是组合产品。位于 https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm 的 510(k) 上市前通知数据库可识别组合产品提交。该法案的一般控制条款包括年度注册、设备清单、良好生产规范、标签以及禁止贴错标签和掺假的要求。请注意:CDRH 不评估与合同责任担保相关的信息。但是,我们提醒您,设备标签必须真实,不得误导。如果您的设备被归类(见上文)为 II 类(特殊控制)或 III 类(PMA),则可能会受到额外控制。现有的影响您设备的重大法规可在《联邦法规》第 21 篇第 800 至 898 部分中找到。此外,FDA 可能会在《联邦公报》上发布有关您设备的进一步公告。请注意,FDA 发布实质等同性认定并不意味着 FDA 已确定您的设备符合该法案的其他要求或其他联邦机构管理的任何联邦法规和规章。您必须遵守该法案的所有要求,包括但不限于:注册和列名(21 CFR 第 807 部分);标签(21 CFR 第

Qure.ai Technologies 2020 年 6 月 17 日 Pooja Rao 负责人......
贸易/设备名称:qER 法规编号:21 CFR 892.2080 法规名称:放射学计算机辅助分类和通知软件 监管类别:II 类 产品代码:QAS 日期:2020 年 6 月 11 日 收到日期:2020 年 6 月 11 日 亲爱的 Pooja Rao: 我们已审查了您根据第 510(k) 条提交的上市前通知,该通知表明您有意销售上述设备,并已确定该设备与在 1976 年 5 月 28 日(医疗器械修正案颁布日期)之前在州际贸易中合法销售的同类设备基本等同(就附件中注明的用途而言),或与已根据《联邦食品药品和化妆品法案》(法案)的规定重新分类的设备基本等同,这些设备不需要获得上市前批准申请(PMA)的批准。因此,您可以营销该设备,但须遵守该法案的一般控制规定。虽然本函将您的产品称为设备,但请注意,一些已获准的产品可能是组合产品。位于 https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm 的 510(k) 上市前通知数据库可识别组合产品提交。该法案的一般控制条款包括年度注册、设备列表、良好生产规范、标签以及禁止贴错标签和掺假的要求。请注意:CDRH 不会评估与合同责任担保相关的信息。但我们提醒您,设备标签必须真实且不得误导。如果您的设备被归类(见上文)为 II 类(特殊控制)或 III 类(PMA),则可能会受到其他控制。影响您设备的现有主要法规可在《联邦法规》第 21 篇第 800 至 898 部分中找到。此外,FDA 可能会在《联邦公报》上发布有关您设备的进一步公告。请注意,FDA 发布实质等效性判定并不意味着 FDA 已判定您的设备符合该法案的其他要求或其他联邦机构管理的任何联邦法规和规章。您必须遵守该法案的所有要求,包括但不限于:注册和列名(21 CFR 第 807 部分);标签(21 CFR 第

2023 年 4 月 3 日 Annalise-AI Pty Ltd. Haylee Bosshard 监管......
贸易/设备名称:Annalise Enterprise CTB Triage Trauma 法规编号:21 CFR 892.2080 法规名称:放射计算机辅助分类和通知软件 监管类别:II 类 产品代码:QAS 日期:2023 年 3 月 1 日 收到日期:2023 年 3 月 1 日 亲爱的 Haylee Bosshard: 我们已审查了您根据第 510(k) 条提交的上市前通知,该通知表明您有意销售上述设备,并已确定该设备与在 1976 年 5 月 28 日(医疗器械修正案颁布日期)之前在州际贸易中合法销售的同类设备基本等同(就附件中所述的使用指征而言),或与根据《联邦食品、药品和化妆品法案》(法案)的规定重新分类的设备基本等同,这些设备不需要获得上市前批准申请(PMA)的批准。因此,您可以营销该设备,但须遵守该法案的一般控制规定。虽然本函将您的产品称为设备,但请注意,一些已获准的产品可能是组合产品。位于 https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm 的 510(k) 上市前通知数据库可识别组合产品提交。该法案的一般控制条款包括年度注册、设备列表、良好生产规范、标签以及禁止贴错标签和掺假的要求。请注意:CDRH 不会评估与合同责任担保相关的信息。但我们提醒您,设备标签必须真实且不得误导。如果您的设备被归类(见上文)为 II 类(特殊控制)或 III 类(PMA),则可能会受到其他控制。影响您设备的现有主要法规可在《联邦法规》第 21 篇第 800 至 898 部分中找到。此外,FDA 可能会在《联邦公报》上发布有关您设备的进一步公告。请注意,FDA 发布实质等效性判定并不意味着 FDA 已判定您的设备符合该法案的其他要求或其他联邦机构管理的任何联邦法规和规章。您必须遵守该法案的所有要求,包括但不限于:注册和列名(21 CFR 第 807 部分);标签(21 CFR 第