XiaoMi-AI文件搜索系统
World File Search System变构

药物化学术语表
附录 2 药物化学术语表 血管紧张素转换酶 (ACE) 抑制剂 一种抗高血压药物,通过抑制血管紧张素转换酶发挥作用,阻止强效血管收缩剂的合成。 乙酰胆碱 (ACh) 神经系统中的一种信使分子。在中枢神经系统中,乙酰胆碱和相关神经元形成胆碱能系统,该系统往往引起抗兴奋作用(另见胆碱能)。 ADMET 指候选药物的吸收、分布、代谢、排泄和毒理学。 激动剂 一种在受体上产生与天然信使相同反应的药物。 变构 指正常配体使用的蛋白质结合位点以外的其他蛋白质结合位点,会影响蛋白质的活性。变构抑制剂与变构结合位点结合会诱导蛋白质形状的改变,从而将正常结合位点与正常配体区分开来。拮抗剂 一种与受体结合但不激活受体的药物,从而抑制天然信使或激动剂的结合。 抗菌剂 一种可以杀死细菌细胞或抑制细菌细胞生长的天然或合成分子。 抗体 一种由人体免疫系统产生的 Y 形糖蛋白,可与外来分子上的抗原相互作用。标记要摧毁的外来分子。 抗体-药物偶联物 一种抗体,其结构与药物共价结合。 抗原 被免疫系统“识别”并与针对它的抗体相互作用的分子区域。 抗代谢物 一种对细胞正常代谢至关重要的酶的抑制剂。用于抗菌和抗癌。 β 受体阻滞剂 一种阻断或拮抗 β 肾上腺素受体的药物。用于心血管方面。 生物测定 一种测量物质对生物体影响的测定方法。生物利用度 给药后,在血浆或靶组织中可利用的药物或其他物质的比例或百分比。 生物标志物 一种生物状态指标,可以可靠地测量和评估,作为生物过程或治疗干预反应的指标。 黑框警告 药品标签上必须出现的最严重的安全警告,表示药物可能出现严重甚至危及生命的不良反应。 血脑屏障 脑血管比周围血管的孔隙率低,且有一层脂肪涂层。针对脑部的药物必须是亲脂性的才能穿过血脑屏障。 化学介导毒性 由于某种化学物质或整个化学物质类别的物理和化学性质而导致的毒性。 胆碱能受体 由乙酰胆碱激活的受体。 慢性粒细胞白血病 一种以髓系细胞过度增殖为特征的血液系统癌症。临床试验第 1 阶段 首先在 50-200 名健康志愿者中测试药物,以确定合适的剂量水平、评估其药代动力学并确定副作用。 临床试验第 2 阶段 在此阶段,在患有目标疾病的患者组(100-500 人)中测试药物,以验证其治疗效果。不同的组接受不同的剂量,通常在双盲条件下进行。 临床试验第 3 阶段 与第 II 阶段类似,但患者人数较多(1000-5000 人)。在此阶段,将证明和充分评估药物的有益效果或其他效果。 临床试验第 4 阶段 在药物获批和上市后,监测其性能是一个永无止境的过程,现在称为第 IV 阶段研究。可能会观察到新的副作用,或者通过长期统计数据揭示对特定群体(例如儿童或孕妇)的影响。如有必要,可以撤回药物。 CNS 中枢神经系统

工程回试产生与基因组无关的蛋白质...
抽象的DNA-蛋白质相互作用是无数天然和合成基因网络的核心组成部分。尽管有潜在的新设计空间,但由于控制特定的DNA片段(包括蛋白质结合序列),DNA-蛋白相互作用在体内仍然没有被体内plaper绕。在这里,我们设计了探针,原核生物的重新元素,以细胞内生成基因组独立的可编程小型DNA,以用于序列特异性蛋白质结合。使用重编程的后衍生的DNA用于变构转录因子,我们证明了合成基因网络的动态调节以及自动反馈电路的构建,以进行信号放大,适应和记忆。此外,我们开发了一种新的刺激反应性分子“诱饵和猎物”,从而使蛋白质亚细胞定位的模块化,快速和翻译后控制能力。这项工作大大扩展了DNA-蛋白质相互作用的可能应用领域,为合成生物学的技术进步奠定了基础。
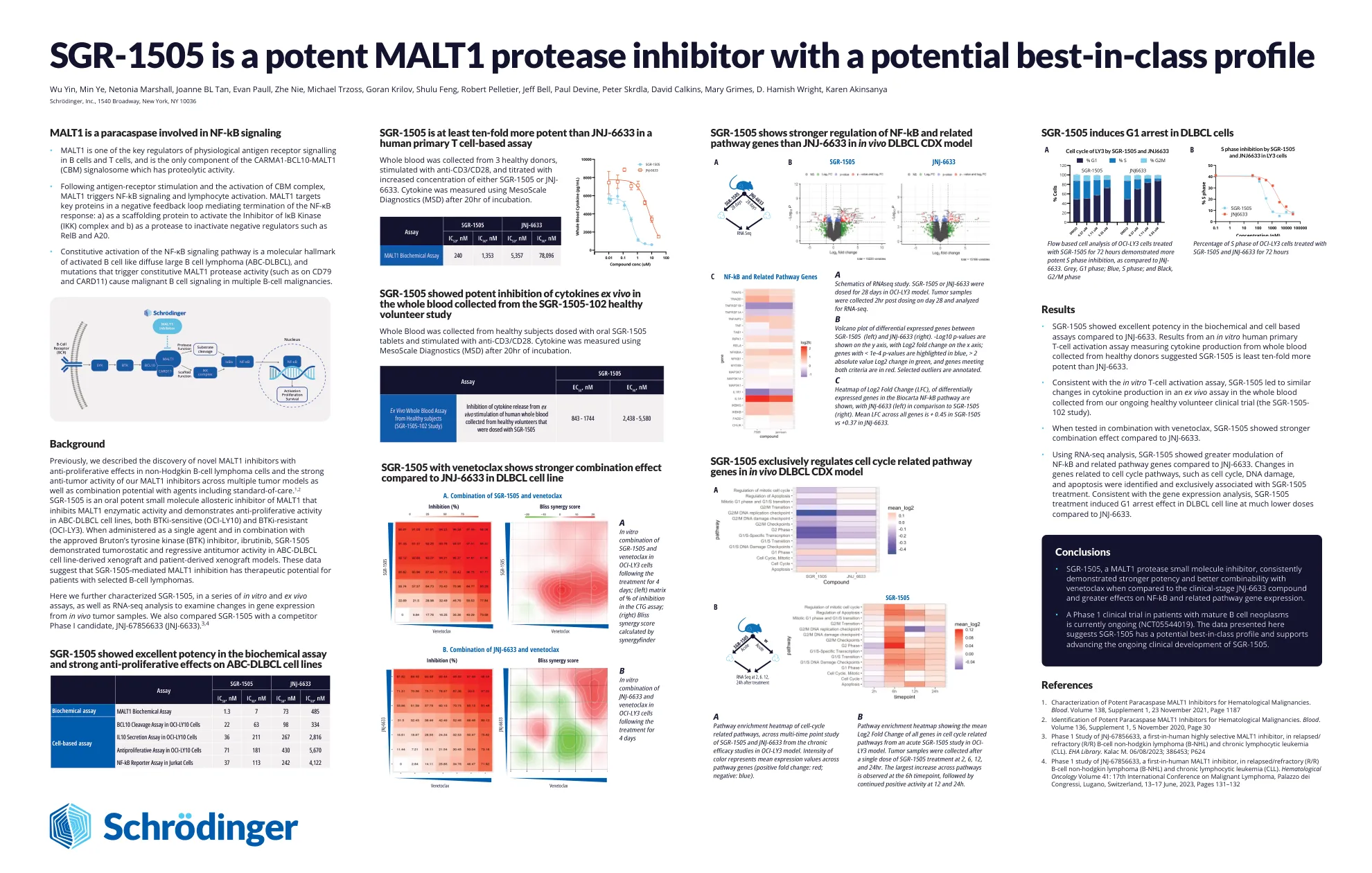
SGR-1505是一种有效的MALT1蛋白酶抑制剂,具有潜在的一流概况
先前,我们描述了在非霍奇金B细胞淋巴瘤细胞中具有抗增殖作用的新型MALT1抑制剂的发现,以及在多个肿瘤模型中MALT1抑制剂的强抗肿瘤活性,以及与包括标准护理在内的药物的组合。1,2 SGR-1505是MALT1的口服有效的小分子变构抑制剂,可抑制MALT1酶活性,并在ABC-DLBCL细胞系中表现出抗增殖活性,均为BTKI敏感性(OCI-LY10)(OCI-LY10)和BTKI-RESIS耐药(OCI-Ly3)。当用作单一药物并与批准的布鲁顿酪氨酸激酶(BTK)抑制剂相结合时,ibrutinib ibrutinib sgr-1505显示出在ABC-DLBCL细胞线衍生的Xenograft中表现出肿瘤抑制和回归抗肿瘤活性。这些数据表明,SGR-1505介导的MALT1抑制作用对于选定的B细胞淋巴瘤患者具有治疗潜力。

LF101-12 生物化学家的物理化学
质量作用定律、速率和平衡 速率常数和反应级数 速率定律和反应机理(零级、一级、二级反应和分数级) 碰撞理论、过渡态理论和阿伦尼乌斯方程 稳态近似 测量反应动力学和确定速率常数的方法,动力学机制建模 酶动力学(米氏动力学、抑制、变构酶;代谢中的酶反应) 影响反应速率的因素(反应的温度依赖性和活化参数、粘度和分子动力学、反应的扩散控制) 复杂反应的动力学分析(瞬态和反应序列研究简介;电子转移和自由基反应动力学;聚合动力学) 生物分子反应动力学和分子药理学简介(蛋白质 - 配体结合和交换动力学;结合位点、单位点和多个独立位点模型、与膜受体结合、降维)

了解药物受体相互作用:解锁药物作用的秘密
药物受体相互作用在药理学领域起关键作用,为理解药物如何发挥其治疗作用和人体内部副作用构成基础。这种复杂的相互作用涉及在细胞表面或细胞内存在的药物分子与特定受体的结合。结合事件会触发一系列分子事件,这些级联可能导致细胞功能改变,信号转导途径和生理反应。本文概述了药物受体相互作用的基本概念,强调了影响结合动力学,亲和力和选择性的关键因素。各种类型的药物相互作用,包括激动剂和拮抗剂相互作用,变构调节和偏置信号传导。此外,强调了解药物发现,开发和个性化医学中药物受体相互作用的重要性。计算建模和结构生物学的进步为这些相互作用的分子机制提供了见解,从而实现了合理的药物设计和优化。通过深入研究药物相互作用的复杂性,研究人员可以在最大程度地减少不良反应的同时获得优化药物疗效的宝贵见解。

二恶英不仅与AHR结合,还与
免疫毒性和内分泌干扰。在1970年代中期,科学家确定了一种被称为芳基烃受体(AHR)的转录因子,该因子随着二恶英的结合而被激活。ahr策划了Nuber的适应性和不良压力反应,并且据信介导了二恶英和DLC触发的大部分(如果不是全部)的毒性作用。最近的研究提供了越来越多的证据,表明二恶英和二恶英样多氯联苯可以通过直接与其细胞外域结合,可以抑制生长因子诱导的表皮生长因子受体(EGFR)的激活。这种相互作用可防止通过聚肽生长因子和下游信号转导的EGFR激活。在本文中,我们详细说明了这种新确定的二恶英和DLC的动作机制,并通过使用两个示例(即乳腺癌发育和胎盘毒性。最后,我们简要介绍了其他全球关注的环境化学物质,这些化学物质基于首次发布的数据,可以通过相同的行动方式起作用。关键词:芳基烃受体,表皮生长因子受体,变构抑制,持续性有机污染物,乳腺癌,胎盘毒性

BTK 活性位点抑制剂对全长 BTK 构象状态的不同影响
摘要布鲁顿酪氨酸激酶 (BTK) 是治疗 B 细胞疾病(包括白血病和淋巴瘤)的靶向药物。目前已获批准的 BTK 抑制剂,包括伊布替尼(一种首创的 BTK 共价抑制剂),可直接与激酶活性位点结合。虽然药物结合可有效阻断 BTK 的催化活性,但药物结合对全长 BTK 整体构象的影响尚不清楚。在这里,我们发现了一组活性位点抑制剂在全长 BTK 中引起的一系列构象效应,包括调节域构象平衡的大规模转变。此外,我们发现 BTK SH2 结构域中的远程伊布替尼抗性突变 T316A 通过破坏全长 BTK 的紧凑自抑制构象来驱动虚假的 BTK 活性,使构象集合远离自抑制形式。 BTK 抑制剂的未来开发将需要考虑抑制剂结合的长期变构后果,包括这些 BTK 抑制剂在治疗 COVID-19 中的新兴应用。

双MEK抑制剂IMM-1- ...
简介:在皮肤,胰腺,肺和结肠中晚期实体瘤的患者中,RAS或RAF中的激活突变是常见的致癌事件。这些患者的治疗选择有限。在打捞环境中,NRAS突变转移性黑色素瘤中的中位总生存期不到一年。MEK位于Ras和Raf的下游,但ERK上游是一个有吸引力的目标,可以抵抗高架MAPK信号传导。虽然MEK抑制剂具有选择性,但FDA注册的MEK抑制剂对RAS突变肿瘤的途径重新激活敏感。这种机械限制促使慢性途径抑制策略有助于限制临床实用性的靶向类别效应的毒性。因此,我们为MEK抑制开发了一种新方法。IMM-1-104是一种新型的变构双MEK抑制剂,可破坏MEK及其下游靶向ERK的磷酸化,并具有短血浆药物半寿命,可通过接近零药槽进行深层循环抑制。

DNA回旋酶中的能量耦合和作用机理
抽象的大肠杆菌DNA速酶催化封闭的双链DNA的否定性超涂层,以ATP为代价。酶的酶的另外活性阐明了超涂层反应的能量偶联成分是ATP至ADP和ADP和PI的DNA依赖性水解,以及ATP通过gyrase裂解反应的DNA位点特异性的ATP改变。这两种DNA链的这种裂解是由稳定的Gy- Rase-DNA复合物的十二烷基硫酸钠处理的,该配合物被抑制剂氧甲酸捕获。ATP或不可水解的类似物,腺基-5'-二氨基磷酸酯(APP [NHLP),都会在Colel DNA上移动主要的裂解位点。这种切割重排的Novobiocin和Coumermycin al的预防将抗生素的作用位点放置在ATP水解之前的一个反应步骤中。步骤阻塞是ATP的结合,因为香豆素和Novobiocin在ATPase和SuperCoiling分析中与ATP竞争相互作用。 K;对于ATP而言,值比KM少四个数量级以上。这种简单的机制解释了药物对DNA回旋酶的所有影响。使用APP [NHP [NHP的另一种有效的反应竞争抑制剂催化YGYRASE的竞争抑制剂,表明将DNA驱动到更高的能量超胶结形式不需要高能键的裂解。 与Gyrase,App的底物水平(NHLP诱导与酶量成正比的超串联; a -0.3超螺旋转弯是根据Gyrase Frotomer A引入的。 我们假设ATP和APP [NH] P是回旋酶的构象变化的变构效应器,导致一轮超涂层。使用APP [NHP [NHP的另一种有效的反应竞争抑制剂催化YGYRASE的竞争抑制剂,表明将DNA驱动到更高的能量超胶结形式不需要高能键的裂解。与Gyrase,App的底物水平(NHLP诱导与酶量成正比的超串联; a -0.3超螺旋转弯是根据Gyrase Frotomer A引入的。我们假设ATP和APP [NH] P是回旋酶的构象变化的变构效应器,导致一轮超涂层。通过ATP水解的核苷酸解离,将回旋酶返回其原始构型,从而允许酶转移。伴随核苷酸亲和力改变的这种环状构象变化似乎也是其他多种操作中能量转导的共同特征,包括肌肉收缩,蛋白质合成和氧化磷酸化。

Aficamten 是一种小分子心脏肌球蛋白抑制剂,用于治疗肥厚型心肌病
肥厚性心肌病 (HCM) 是一种遗传性肌节疾病,会导致心脏收缩过度。一流的心脏肌球蛋白抑制剂 mavacamten 可改善阻塞性 HCM 的症状。我们在此介绍一种选择性小分子心脏肌球蛋白抑制剂阿菲卡汀,它通过显著减缓磷酸盐释放来降低 ATPase 活性,从而稳定弱肌动蛋白结合状态。阿菲卡汀与肌球蛋白催化域上的变构位点结合,不同于 mavacamten,可防止进入强肌动蛋白结合力产生状态所需的构象变化。通过这样做,阿菲卡汀减少了驱动肌节缩短的功能性肌球蛋白头部的数量。在前动力冲刺状态下与心脏肌球蛋白结合的阿菲卡汀的晶体结构为理解其对平滑肌和快速骨骼肌的选择性提供了基础。此外,在心肌细胞和携带肥大性 R403Q 心肌肌球蛋白突变的小鼠中,阿菲卡汀可降低心脏收缩力。我们的研究结果表明,阿菲卡汀有望成为 HCM 的治疗方法。