XiaoMi-AI文件搜索系统
World File Search System外切

抑制α-葡萄糖苷酶活性的机制...
α-葡萄糖苷酶(EC 3.2.1.20)是一种碳水化合物水解酶,广泛分布于小肠黏膜刷状缘,对糖基结构有重要影响。它能以内切或外切的方式水解各种糖化合物中的糖苷键,产生单糖、寡糖或糖胺聚糖,导致餐后血糖升高(Daub et al., 2020; Ismail et al., 2020; Attjioui et al., 2020)。餐后高血糖是导致2型糖尿病发生、发展的主要危险因素。抑制α-葡萄糖苷酶活性可减慢碳水化合物的消化,从而减少葡萄糖吸收入血,控制血糖水平。这种抑制被认为是治疗非胰岛素依赖型糖尿病的重要临床验证靶点(Ye et al., 2019; Khan et al., 2019; Syabana et al., 2021)。目前常用的α-葡萄糖苷酶抑制剂为阿卡波糖、伏格列波糖等生物合成或半生物合成药物,这些药物价格昂贵,且有不同程度的不良副作用(主要为腹部不适、恶心、呕吐等胃肠道反应(Wehmeier & Piepersberg, 2004; Smith et al., 2021)。需要开发安全、有效、具有临床获益的新型α-葡萄糖苷酶抑制剂。
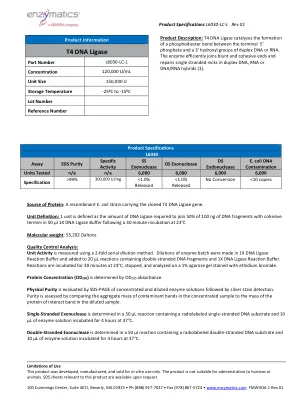
T4 DNA连接酶
蛋白质的来源:带有克隆的T4 DNA连接酶基因的重组大肠杆菌菌株。单位定义:1个单位定义为将100 ng的DNA片段中的50%与粘性末端连接到50 µl 1x 1x DNA连接酶缓冲液后30分钟在23°C分子重量下孵育后所需的50%的DNA片段:55,292 DALTONS质量控制分析:使用2ffliutial serial dilitial doldutial doldiques soge。在1x DNA连接酶反应缓冲液中制作酶批次的稀释液,并添加到含有双束DNA片段和1X DNA连接酶反应缓冲液的20 µL反应中。在23°C下孵育30分钟,停止并在用溴化乙锭染色的1%琼脂糖凝胶上进行分析。蛋白浓度(OD 280)由OD 280吸光度确定。物理纯度,然后进行银色染色检测。通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。单链核酸酶在含有放射性标记的单链DNA底物的50 µL反应中确定,在37°C下孵育4小时4小时。双链外切核酸酶在50 µL反应中确定,该反应含有放射性标记的双链DNA底物和10 µL的酶溶液在37°C下孵育4小时。

TAQ-B DNA聚合酶
蛋白质的来源:一种重组大肠杆菌菌株,携带来自嗜热有机体Thermus aquaticus YT-1的TAQ DNA聚合酶基因。单位定义:1个单位定义为将在75°C的30分钟内将10 nmol的DNTP纳入酸 - 不溶性材料的酶。分子量:93,910 Daltons质量控制分析:使用2倍连续稀释方法测量单位活动。在1X反应缓冲液中制成酶的稀释液,并将其添加到含有小腿胸腺DNA,25 mM TAPS(pH 9.3),50 mM KCl,2.0mm MGCL2,1 mM DTT,3H-DTTP和100 µm DNTP的50 µL反应中。 在75°C下孵育10分钟,浸入冰上,并使用Sambrook和Russell的方法进行分析(Molecular Cloning,V3,2001,pp。 A8.25-A8.26)。 蛋白浓度(OD 280)由OD 280吸光度确定。 通过浓缩和稀释酶溶液的SDS-PAGE评估物理纯度,然后进行银色染色检测。 通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。 单链核酸酶在含有放射性标记的单链DNA底物的50 µL反应中确定,在37°C下孵育4小时4小时。 双链外切核酸酶在50 µL反应中确定,该反应含有放射性标记的双链DNA底物和10 µL的酶溶液在37°C下孵育4小时。 双链核酸内切酶在50 µL反应中确定,该反应含有0.5 µg质粒DNA和10 µL的酶溶液在37°C下孵育4小时。在1X反应缓冲液中制成酶的稀释液,并将其添加到含有小腿胸腺DNA,25 mM TAPS(pH 9.3),50 mM KCl,2.0mm MGCL2,1 mM DTT,3H-DTTP和100 µm DNTP的50 µL反应中。在75°C下孵育10分钟,浸入冰上,并使用Sambrook和Russell的方法进行分析(Molecular Cloning,V3,2001,pp。A8.25-A8.26)。蛋白浓度(OD 280)由OD 280吸光度确定。物理纯度,然后进行银色染色检测。通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。单链核酸酶在含有放射性标记的单链DNA底物的50 µL反应中确定,在37°C下孵育4小时4小时。双链外切核酸酶在50 µL反应中确定,该反应含有放射性标记的双链DNA底物和10 µL的酶溶液在37°C下孵育4小时。双链核酸内切酶在50 µL反应中确定,该反应含有0.5 µg质粒DNA和10 µL的酶溶液在37°C下孵育4小时。
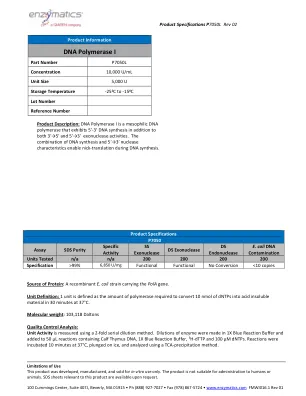
DNA聚合酶I
蛋白浓度(OD 280)由OD 280吸光度确定。物理纯度,然后进行银色染色检测。通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。单链核酸酶在含有放射性标记的单链DNA底物的50 µL反应中确定,在37°C下孵育4小时4小时。双链外切核酸酶在50 µL反应中确定,该反应含有放射性标记的双链DNA底物和10 µL的酶溶液在37°C下孵育4小时。双链核酸内切酶在50 µL反应中确定,该反应含有0.5 µg质粒DNA和10 µL的酶溶液在37°C下孵育4小时。e.coli 16S rDNA污染使用5 µL R含量的酶溶液的样品变性并在Taqman QPCR测定中筛选,以使用污染的大肠杆菌基因组DNA,使用寡核苷酸引物污染了与16S rRNA locus相对应的寡核苷酸引物。提供:25mm Tris-HCl,1mm DTT,0.1mm EDTA,50%甘油(25°C时pH 7.4)。提供:10倍蓝色缓冲区(B0110):500mm NaCl,100mm Tris-HCl,100mm MGCL 2,10mm DTT(25 c)pH 7.9 pH 7.9)。用法说明:5´-overhang(1)

G2 DNA/RNA增强子
G2 DNA/RNA增强子可以方便地使用,尤其是尤其是粘土中需要最佳的DNA和/或RNA提取产率时。G2 DNA/RNA增强子的主要功能是减轻抑制性DNA-粘土颗粒的形成。G2 DNA/RNA增强子增加了粘土的微生物DNA和RNA产量 - 至少2-10倍。G2 DNA/RNA增强剂应与标准化提取方法或用于从土壤和粘土中提取DNA和RNA的商业试剂盒结合使用。建议在-20至25°C处进行存储和稳定性存储。保持干燥。质量控制G2 DNA/RNA增强子进行污染活性,没有核酸内核酸酶活性,缺口活性,外切核酸酶活性或RNase活性的痕迹。此外,在难以提取的矩阵中,对G2 DNA/RNA增强子进行了功能测试。套件组件Ampliqon G2 DNA/RNA增强子冻结干燥的G2 DNA/RNA增强剂和2 mL管中的1.4 mM珠。协议使用G2 DNA/RNA增强子时,该方案是DNA和RNA提取的指南。G2 DNA/RNA增强子必须使用提取套件施加。程序:将0.25克土壤样品添加到G2 DNA/RNA增强器管中。应用您的DNA或RNA隔离套件。例如Dneasy Powersoil Pro Kit。o如果套件的珠珠管中包含裂解缓冲液,请将此裂解缓冲液转移到G2管上,并丢弃现在空的套件的珠珠管。

5'-磷酸盐增强了人线粒体基因组维持外核酸酶1(MGME1)
在较高的真核生物中,线粒体在能量生产,信号传导和生物合成中起多种作用。线粒体具有多个线粒体DNA(mtDNA)的副本,该线粒体DNA(mtDNA)编码了37个对于线粒体和细胞功能必不可少的基因。当mtDNA受到内在和外源性因素和外源性因素的挑战时,MTDNA经历修复,降解和补偿性合成。mtDNA降解是mtDNA损伤响应和维持中的新兴途径。涉及的关键因素是人线性基因组维持外切酶1(MGME1)。尽管以前的生化和功能研究,但关于MGME1介导的DNA裂解的极性存在争议。此外,DNA序列如何影响MGME1的活性仍然难以捉摸。这种信息不仅是对MGME1的理解的基础,而且对于决定mtDNA降解机制至关重要。在此,我们使用定量测定来检查底物结构和序列对MGME1的DNA结合和酶促活性的影响。我们证明了MGME1与单链DNA底物的5 0端结合并切割,尤其是在5 0-磷酸盐存在下,在DNA结合和MGME1的最佳裂解中起重要作用。此外,MGME1在末端耐受某些修饰,例如在基础切除修复中形成的5 0-脱氧核糖磷酸磷酸盐中间体。我们表明,MGME1通过不同的效率处理不同的序列,而DT和DC序列分别是最多,有效地消化的序列。我们的结果提供了对MGME1的酶促特性的见解,以及MGME1与MTDNA降解中DNA聚合酶γ的3 0 - 5 0外核酸酶活性的配位基本原理。

rNase抑制剂
蛋白质的特异性RNase源:带有猪RNase抑制剂基因的重组大肠杆菌菌株。单位定义:1个单位定义为抑制50%cytidine 2',3'-循环单磷酸的水解所需的酶量,由5 ng的RNase A(1)抑制。分子量:74,828 Daltons质量控制分析:使用2倍连续性稀释方法确定单位活动。在1x RNase抑制剂反应缓冲液中制成酶的稀释液,并添加到含有1mm胞苷2',3'-环磷酸1mm的1000 µL反应中,其中包含100mm tris-tris-tris-tris-trisate,1mm Edta,1mm Edta,pH 6.5的1X反应缓冲液中的1μgrNase A。在286nm处的吸光度。蛋白浓度(OD 280)由OD 280吸光度的物理纯度确定,通过浓缩和稀释的酶溶液的SDS-PAGE,然后是银色染色检测。通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。单链核酸酶在含有放射性标记的单链DNA底物的50 µL反应中确定,在37°C下孵育4小时4小时。双链外切核酸酶在50 µL反应中确定,该反应含有放射性标记的双链DNA底物和10 µL的酶溶液在37°C下孵育4小时。双链核酸内切酶在50 µL反应中确定,该反应含有0.5 µg质粒DNA和10 µL的酶溶液在37°C下孵育4小时。

通过核酸外切酶融合改进FnCas12a基因组编辑
通过外切酶融合改进 FnCas12a 基因组编辑 吴永强 1*、袁其晨 2*、朱玉峰 3、高翔 4、宋家宝 5、尹子如 6 1 河北科技大学基因编辑研究中心,河北石家庄 050018。2 美国莱斯大学化学与生物分子工程系,休斯顿,德克萨斯州 77005,美国。3 河北科技大学科技发展研究所,河北石家庄。4 河北科技大学环境科学与工程学院,河北石家庄 050018。5 河北科技大学生物科学与生物工程系,河北石家庄。 6 河北科技大学期刊出版社,河北石家庄 050018。 * 通讯作者:吴永强(wuyongqiang@hebust.edu.cn),袁其晨(yqc@rice.edu) 摘要 在目前报道的 Cas12a 直系同源物中,新凶手弗朗西斯菌 Cas12a (FnCas12a) 受原型间隔区相邻基序 (PAM) 的限制较少,这有助于靶向以前无法接近的基因组位点。然而,FnCas12a 核酸酶的活性相对较低或无法观察到,限制了其作为理想基因组工程工具的应用。在这里,我们描述了 TEXT(将 EXonuclease T5 与 FnCas12a 连接),这是一种融合策略,可显着提高 FnCas12a 在人类细胞中在三种不同细胞系中的多个基因组位点的敲除效率。 TEXT 在 18nt 至 23nt 不同间隔长度下均表现出比 FnCas12a 更高的插入和缺失效率,其中 18nt 的插入效率最高,比 FnCas12a 高出 11 倍。深度测序表明 TEXT 显著增加了目标位点的缺失频率和大小。总之,TEXT 增强了 FnCas12a 核酸酶的活性,拓展了其在人类细胞基因组工程中的应用。
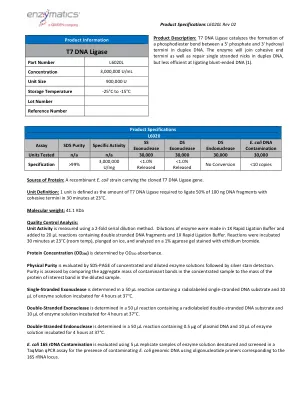
T7 DNA连接酶
蛋白质的来源:一种带有克隆的T7 DNA连接酶基因的重组大肠杆菌菌株。单位定义:1个单位定义为在30分钟内在23°C下在30分钟内将100 ng DNA片段的50%结合的T7 DNA连接酶的量。分子量:41.1 kDa质量控制分析:使用2倍连续稀释方法测量单位活动。稀释液,并添加到含有双链DNA片段和1倍快速连接缓冲液的20 µL反应中。在23°C(室温)下孵育30分钟,浸在冰上,并在用溴化乙锭染色的1%琼脂糖凝胶上进行分析。蛋白浓度(OD 280)由OD 280吸光度确定。物理纯度,然后进行银色染色检测。通过比较浓缩样品中污染物带的聚集质量与稀释样品中蛋白蛋白蛋白带的质量来评估纯度。单链核酸酶在含有放射性标记的单链DNA底物的50 µL反应中确定,在37°C下孵育4小时4小时。双链外切核酸酶在50 µL反应中确定,该反应含有放射性标记的双链DNA底物和10 µL的酶溶液在37°C下孵育4小时。双链核酸内切酶在50 µL反应中确定,该反应含有0.5 µg质粒DNA和10 µL的酶溶液在37°C下孵育4小时。大肠杆菌16S rDNA的污染是使用5 µL r菌酸溶液的样品变性的样品,并在Taqman QPCR分析中筛选,以使用与16S rRNA locus相应的寡核苷酸引物,使用污染的大肠杆菌Genomic DNA。

通过核酸外切酶融合改进FnCas12a基因组编辑
通过外切酶融合改进 FnCas12a 基因组编辑 吴永强 1*,袁其晨 2*,朱玉峰 3,高翔 4,宋家宝 5,尹子如 6 1 河北科技大学基因编辑研究中心,河北石家庄 050018。2 美国莱斯大学化学与生物分子工程系,休斯顿,德克萨斯州 77005,美国。3 河北科技大学科技发展研究所,河北石家庄。4 河北科技大学环境科学与工程学院,河北石家庄 050018。5 河北科技大学生物科学与生物工程系,河北石家庄。 6 河北科技大学期刊出版社,河北石家庄 050018。 * 通讯作者:吴永强(wuyongqiang@hebust.edu.cn),袁其晨(yqc@rice.edu) 摘要 在目前报道的 Cas12a 直系同源物中,新凶手弗朗西斯菌 Cas12a (FnCas12a) 受原型间隔区相邻基序 (PAM) 的限制较少,这将有助于靶向以前无法接近的基因组位点。然而,FnCas12a 核酸酶的活性在人体细胞中相对较低或无法检测到,限制了其作为理想基因组工程工具的应用。在这里,我们描述了 TEXT(将 EXonuclease T5 与 FnCas12a 连接),这是一种融合策略,可显着提高人体细胞中 FnCas12a 在三种不同细胞系中的多个基因组位点的敲除效率。使用18nt至23nt的不同间隔长度,TEXT的插入和删除(indel)效率均高于FnCas12a,其中18nt的插入效率最高,比FnCas12a高出11倍。深度测序表明,TEXT显著增加了目标位点的删除频率和删除大小。TEXT增强了FnCas12a核酸酶的活性,拓展了其在人类细胞基因组工程中的应用。