XiaoMi-AI文件搜索系统
World File Search System富集的

长的非编码RNA DLEU2和ROR1途径在乳腺癌中诱导上皮到间质转变和癌症干细胞
乳腺癌(BC)患者因延长时间而接受化学疗法的患者可能会因上皮到中层过渡(EMT)机制(EMT)机制和富集的癌症干细胞(CSC)而引起的转移和临床结局产生深远的影响。在淋巴细胞性白血病-2(LNCRNA DLEU2)和I型酪氨酸激酶样孤儿受体ROR1(ROR1)中表达高水平LNCRNA的BC细胞可能在激活EMT和CSC诱导的增强能力中起作用。在这里我们发现,与TCGA,PubMed Geo数据集和来自存档的乳腺癌肿瘤组织的样品相比,在肿瘤组织中特异性上调了lncRNA dleu2和ror1。化学疗法后,在BC肿瘤细胞中增强了LNCRNA dleu2和ROR1,并与CSC,EMT相关基因和BMI1的表达结合。机械上,ROR1和LNCRNA DLEU2过表达导致肿瘤细胞增殖增强,抑制凋亡,细胞周期失调,化学耐药性以及BC细胞侵入,迁移,发展球体的能力。这些发现意味着lncRNA dleu2和ror1在BC治疗衰竭中的作用在很大程度上归因于EMT,这与富集的CSC相关。总而言之,我们的发现表明LNCRNA DLEU2和基于ROR1的调节环控制EMT和CSC自我更新,这意味着针对这种调节途径可以改善患者对化学疗法和存活的反应。


有丝分裂原和应激激活蛋白激酶1负调节海马神经发生
摘要 - 成人海马的亚晶体区(SGZ)中的神经发生,可以通过多种手段来刺激,包括通过将实验动物暴露于丰富的环境中,从而提供额外的鼻子,社交和运动刺激。在丰富的动物中产生的有形健康和认知益处,包括改善对精神病,神经学和神经退行性疾病的建模,这可能会影响人类,这可能部分是由于神经元的产生增强所致。神经元反应富集的关键因素是释放脑衍生的神经营养因子(BDNF)和有丝分裂原活化蛋白激酶(MAPK)级联反应的激活,这可能导致刺激Neuroogenese或Neuroogenese的刺激。有丝分裂原和应激激活的蛋白激酶1(MSK1)是BDNF和MAPK下游的一种核酶,可调节转录。MSK1先前已经与缺乏MSK1蛋白的小鼠的研究有关基础和刺激的神经发生。在本研究中,使用仅缺乏MSK1激酶活性的小鼠,我们表明SGZ(KI-67染色)的细胞增殖速率没有由MSK1激酶DEAD(KD)突变造成的,并且与控制后水平的水平没有分歧。然而,与野生型小鼠相比,在标准housed和富集的MSK1 KD小鼠中,双铁蛋白(DCX)阳性细胞的数量都更大。2020年作者。由Elsevier Ltd代表IBRO出版。这是CC BY-NC-ND许可证(http://crea-tivecommons.org/licenses/by-nc-nd/4.0/)下的开放访问文章。这些观察结果表明,尽管MSK1不影响神经元前体的增殖基础速率,但MSK1负责调节注定成为神经元的细胞数量,可能是对新神经元数量的稳态控制,而新神经元的数量则是整合到齿状gyrus中的新神经元的数量。

稳定同位素探测宏基因组学的标准化定量分析策略
抽象稳定的同位素探测(SIP)促进了通过核酸的同位素富集对复杂生态系统中活性微生物种群的培养无关鉴定。许多DNA-SIP研究依赖于16S rRNA基因序列来识别活性分类单群,但是将这些序列与特定细菌基因组联系起来通常具有挑战性。在这里,我们描述了一个标准化的实验室和分析框架,用于使用shot弹枪元基因组学而不是16S rRNA基因测序以人均基因量化同位素富集。为了开发此框架,我们使用设计的微生物组探索了各种样本处理和分析方法,其中标记的基因组的身份及其同位素富集的水平得到了实验控制。使用此基础真理数据集,我们经验评估了不同分析模型的准确性,以识别活性分类单元,并检查了测序深度如何影响同位素标记的基因组的检测。我们还证明,使用合成DNA内部标准来测量SIP密度分数中的绝对基因组丰度可改善同位素富集的估计值。此外,我们的研究说明了内部标准的效用,以揭示样品处理中的异常情况,如果未被发现,可能会对SIP元基因组分析产生负面影响。最后,我们提出了SIPMG,这是一个R软件包,可促进绝对丰度的估计并执行统计分析,以识别SIP元基因组数据中标记的基因组。这个经过实验验证的分析框架增强了DNA-SIP宏基因组学的基础,作为准确测量环境微生物种群的原位活性并评估其基因组潜力的工具。
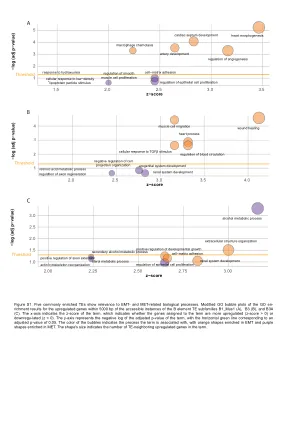
图S1
图S1。 五个通常富集的TE与EMT和MET相关的生物学过程相关。 在B1元素te subfamilies b1_mus1(a),b3(b)和b3a(c)的可访问实例的5000 bp内的GO富集结果的修改后的GO气泡图。 x轴表示该术语的z评分,该术语表明分配给该术语的基因是更上调的(z分数> 0)还是下降的(z <0)。 y轴表示该术语调整后的P值的负log,水平绿线对应于调整后的P值为0.05。 气泡的颜色表示该术语与之相关的过程,橙色形状富含EMT和紫色形状富含MET。 形状的大小指示了该术语中上调的基因的数量。图S1。五个通常富集的TE与EMT和MET相关的生物学过程相关。在B1元素te subfamilies b1_mus1(a),b3(b)和b3a(c)的可访问实例的5000 bp内的GO富集结果的修改后的GO气泡图。x轴表示该术语的z评分,该术语表明分配给该术语的基因是更上调的(z分数> 0)还是下降的(z <0)。y轴表示该术语调整后的P值的负log,水平绿线对应于调整后的P值为0.05。气泡的颜色表示该术语与之相关的过程,橙色形状富含EMT和紫色形状富含MET。形状的大小指示了该术语中上调的基因的数量。

Biomek Ngenius Illumina Trusight肿瘤学500 DNA仅自动化套件应用程序模板设置指南
Illumina Trusight肿瘤学500 DNA自动化套件应用DNA仅允许创建与Illumina测序平台兼容的库。f it tollowing批处理,可以将Covaris剪切的DNA样品加载到库制备反应容器(RV)上,并通过末端修复/A-tailing,适配器连接和索引PCR制备到Illumina库中。感兴趣的区域与探针杂交,磁捕获和洗脱,并且富集的文库被杂乱无章。可选的荧光定量步骤可用于确保在基于珠子的归一化之前有足够的库。归一化后,库准备池进行测序。下面的图1详细介绍了工作流的特定自动化和手动步骤。

B7H3、CD22、PTK7 和 CD33
2023 年 9 月,COACH 召开了第六次会议,与会代表包括美国国家癌症研究所 (NCI)、美国食品药品管理局 (FDA)、欧洲药品管理局 (EMA)、倡导团体、制药行业、儿科临床前概念验证平台 (ITCC-P4) 和儿科临床前体内测试 (PIVOT) 联盟,旨在评估四个癌症相关靶点,这些靶点可能是开发用于儿科适应症的救命药物的潜在实体。具体来说,这次会议重点关注了癌细胞表面富集的靶点,从而能够更直接、更具体的方式将细胞毒性疗法递送至肿瘤。

寄生虫负荷对绦虫及其蚂蚁中间宿主转录活性和形态的影响
图 2 对 122 个蚂蚁差异表达基因中的 120 个进行聚类和可视化。根据基因的表达模式,可将其分为三个簇:(a)簇 1、(b)簇 2 和(c)簇 3。使用 topGO 和 weight01 算法计算这些簇的 GO 富集分析(簇 1 为 d、簇 2 为 e、簇 3 为 f),并使用 Fisher 精确检验将簇的生物学过程的 GO 注释与整个转录组进行比较。每个条形图代表每个簇中显著富集的 GO 术语,x 轴代表显著基因的数量。

论文摘要
表格列表 表格 页码 表 2.1. 根据 Sandvik 数据表的粉末化学成分…………………………………………………………………………………….. 22 表 2.2. 本研究使用的优化 LDED 工艺参数……………………………….. 23 表 2.3. 316LY 原料粉末的物理性质……………………………………..25 表 2.4. 打印状态和热稳定性测试的 316LY ODS 中富集的氧化物纳米颗粒的 EDS 化学分析…………………………………………………………31 表 2.5. 打印状态的 LDED 316LY ODS 中的晶粒尺寸与在 1000 ℃ 下 100 小时后的晶粒尺寸比较……………………………………………………………….33 表 2.6. 采用不同生产工艺生产的样品的机械性能比较…………………………………………………..34 表 2.7.对打印和热老化后的 LDED 316LY 700W 凹坑进行 EDS 点分析化学分析 ………………………………………………… 37

IDT高通量杂交捕获术的长基因组片段的富集证明了协议(RUO23-2352_001 09/23)
图1。高通量杂交捕获量的长基因组片段工作流程。 (a)高分子量(HMW)基因组DNA需要长片段的制备和富集。 (b)使用高通量兼容的G管将HMW DNA碎片至〜10 kb。 (c)使用特殊准备的尺寸选择珠通过尺寸选择去除多余的较小片段。 (d)尺寸选定的片段被最终修复(ER)A-Tail(AT),并将适配器连接到适配器序列,并带有样品识别条形码序列。 (e)样品池与XGEN自定义HYB面板杂交,并捕获并富集。 (f)富集的目标片段通过远程PCR扩增。 (g)放大的富集片段是第二次尺寸选择的或清理以进行最佳测序读取长度。 然后,富集和条形码的样品池接受所需的第三代测序工作流程。高通量杂交捕获量的长基因组片段工作流程。(a)高分子量(HMW)基因组DNA需要长片段的制备和富集。(b)使用高通量兼容的G管将HMW DNA碎片至〜10 kb。(c)使用特殊准备的尺寸选择珠通过尺寸选择去除多余的较小片段。(d)尺寸选定的片段被最终修复(ER)A-Tail(AT),并将适配器连接到适配器序列,并带有样品识别条形码序列。(e)样品池与XGEN自定义HYB面板杂交,并捕获并富集。(f)富集的目标片段通过远程PCR扩增。(g)放大的富集片段是第二次尺寸选择的或清理以进行最佳测序读取长度。富集和条形码的样品池接受所需的第三代测序工作流程。