XiaoMi-AI文件搜索系统
World File Search System序列

基于CRISPR/Cas9基因编辑技术的人类遗传疾病 ...
摘要 CRISPR/Cas9 系统 ( 常间回文重复序列丛集 / 常间回文重复序列丛集关联蛋白系统 ) 为靶向基因编辑提 供了强大的技术手段 . 利用序列特异性 sgRNA 的引导 , CRISPR/Cas9 系统能够精准地在目标 DNA 的确切位置导 入双链切口 . 与已有的基因编辑手段相比 , 该系统具有更优异的简便性、特异性和有效性 . 目前 , 大量涉及体内 外多物种的 CRISPR/Cas9 基因编辑研究已充分展示了该技术的巨大潜力 , 为基于该技术的疾病治疗研究和临床 应用带来了希望 . 基于 CRISPR/Cas9 基因编辑技术所介导的非同源性末端连接和同源性 DNA 修复作用 , 近期多 个研究工作已经成功应用该技术修复了包括点突变和基因组缺失等在内的遗传疾病相关基因组缺陷 . 本综述 将总结近期有关利用 CRISPR/Cas9 基因编辑技术治疗人类遗传性疾病的相关临床前研究进展 .

2024 年课程序列手册
• CHI 201 中级汉语 I • CHI 202 中级汉语 II • FRE 201 中级法语 I • FRE 202 中级法语 II • JPN 201 中级日语 I • JPN 202 中级日语 II • LAT 201 中级拉丁语 I • LAT 202 中级拉丁语 II • POR201 中级葡萄牙语 I • POR 202 中级葡萄牙语 II • SPA201 中级西班牙语 I • SPA202 中级西班牙语 II • ASL 201 美国手语 I • ASL202 美国手语 II 学生必须以最低“D”的成绩完成 202 才能满足核心课程要求。学生可以完成世界语言文学系批准的其他语言。

越南战争的战斗欺骗序列:1965-68
本论文是在候选人的论文导师、历史系的约翰·奥沙利文博士的指导下准备的,并已获得其监督委员会成员的批准。它已提交给艺术与人文学院的教职员工,并被接受为部分满足文学硕士学位的要求。
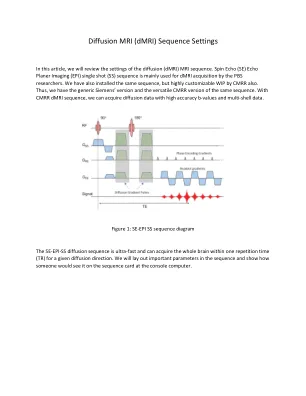
扩散 MRI (dMRI) 序列设置
GRAPPA 是平面内加速因子;GRAPPA 为 2 时,扫描时间将减少近一半。多波段因子 (SMA) 是切片加速因子;SMA 为 4 时,扫描时间将减少近四分之一。部分傅立叶沿相位编码方向削减一些 k 空间线以加速采集;如果使用,大多数研究使用 6/8 因子。如果需要,我们可以同时应用所有这些加速技术,但会牺牲图像质量。大多数研究使用 GRAPPA 为 2 和 SMA 为 2 或 4。一些研究人员只使用 SMA 为 8。使用 SMA,一些研究人员还保存参考扫描以供后期处理。

全体形态序列的边界动力学,非...
我们考虑了全体形态映射的序列f n:u→u→u→n∈N,在正确的,简单地在复杂平面c的相互连接的子域之间,并证明了有关序列(f n(z))之间关系的结果,其中z是u和(f n(f n(ζζ))的内部点,位于U边界中。通常,映射f n并未在u的边界上定义,但是我们将引入f n的“径向扩展”,在许多情况下,对于谐波措施,在许多情况下,在许多情况下存在,在这种情况下 - 在这种情况下,我们说f n具有“完全径向扩展”到∂u;有关详细信息,请参见第2节。为简单起见,我们使用符号f n也参考了在这些边界点处的原始地图的扩展。

mRNA序列参数和公式优化...
▪129 sV小鼠▪tlr7/8铅拮抗剂与mRNA以各种比率混合▪编码萤火虫荧光素酶的未经改性mRNA▪拮抗剂 - 拮抗剂 - 液化液作用为LNP▪静脉注射10或30 µg mRNA-LNP的静脉内注射和6H和24H分析的ERNAINS INSTER ERNAINS INSTER ERNAINS ERANTION ERANTION ERANTION ERANTION ERANTION ERANTION ERANTION ERANTION ERANTION ERANTION + ERNAINS分析▪从血清▪器官裂解物中的荧光素酶活性

索引所有人生的已知生物序列
(‡等等贡献。∗应向谁解决。)9公共存储库中可用的生物测序数据量正在成倍增长,形成了10个宝贵的生物医学研究资源。然而,使其在11种生活和数据科学中的研究人员可以访问且易于访问是一个未解决的问题。在这项工作中,我们利用了最近开发的,非常有效的12个数据结构和算法来表示序列集。我们在所有13个生命的进化枝中制作了DNA序列的石柄,包括病毒,细菌,真菌,植物,动物和人类,都可以完全搜索。我们的索引可供研究社区免费使用。在单个消费者硬盘驱动器(≈100USD)上,输入序列(最多15 5800×)的高度压缩表示形式,使使用可使用的有价值的资源成本效益和16个易于运输。我们提出了一种基本的方法论框架,称为Metagraph,该框架使我们使用注释的DE Bruijn图可缩减索引非常大的DNA或蛋白质序列。我们证明了18个可行性,即索引现有的测序数据的全部范围,并提出新的方法,以实现高效和成本-19有效的全文搜索,按点数为0.10美元,每个查询的MPB $ 0.10。我们探索了几个实际用例20,以挖掘现有的档案,以进行有趣的关联,并证明了我们对综合21分析的索引的实用性。22

中国社会的序列生成对抗网络...
尽管序列到序列模型在许多摘要数据集中都达到了最先进的性能,但是中国社交媒体文本的处理中仍然存在一些问题,例如短句子,缺乏连贯性和准确性。这些问题是由两个因素引起的:基于RNN的序列模型的原理是最大似然估计,这将导致产生长摘要时梯度消失或爆炸;中国社交媒体中的文字漫长而嘈杂,很难产生高质量的摘要。为了解决这些问题,我们应用了序列生成的对抗网络框架。该框架包括生成器和歧视器,其中生成器用于生成摘要,并使用歧视器来评估生成的摘要。软核层层用作连接层,以确保发电机和歧视器的共培训。实验是在大规模的中国社交媒体文本摘要数据集上进行的。句子的长度,胭脂评分和摘要质量的人工得分用于评估生成的摘要。结果表明,我们的模型生成的摘要中的句子更长且准确性更高。

使用奴才序列对SARS-COV-2分析
我们将要使用的数据是武汉海鲜市场肺炎爆发的元文字小奴才数据集[Chan等,2020]。对疾病的病毒进行采样涉及从患者那里获得痰,喉咙或鼻咽拭子或支气管肺泡灌洗液(BALF)样品。因此,样品将包含来自非病毒源的RNA。该数据集是使用独立于序列的单播放扩增(SISPA)方案来制备的,以进行其他病毒序列富集。

DNA序列生成的潜在扩散模型
机器学习的利用,尤其是深层生成模型,已在合成DNA序列产生的领域开辟了有希望的途径。虽然生成对抗网络(GAN)在此应用中获得了吸引力,但他们经常面临诸如样本多样性和模式崩溃等问题。另一方面,扩散模型是一种有希望的新的生成模型,这些模型不承担这些问题的负担,从而使它们能够达到图像生成等领域的最先进。鉴于此,我们提出了一种新型潜在扩散模型,即用于离散DNA序列产生的新型潜扩散模型。通过使用自动编码器将离散的DNA序列简单地嵌入连续的潜在空间中,我们能够利用连续扩散模型的强大生成能力来生成离散数据。此外,我们将Fréchet重建距离(FRED)作为新指标,以测量DNA序列世代的样品质量。我们的码头模型表明,就基序分布,潜在嵌入分布(FRED)和染色质曲线而言,与实际DNA紧密对齐的合成DNA序列具有能力。此外,我们还提供了来自15种的150K独特启动子基因序列的综合跨物种数据集,丰富了基因组学中未来生成建模的资源。我们已在https://github.com/zehui127/latent-dna-diffusion上公开提供代码和数据。