XiaoMi-AI文件搜索系统
World File Search System种树

基因1表达在野生西红柿2
现在可以理解,渗入可以充当强大的进化力,提供了31种遗传变异,可以影响性状进化的过程。渗入还引起了共有的32个进化史,该历史不会被物种系统发育所捕获,可能会使使用物种树的33个进化分析复杂化。这种分析通常是对跨物种的基因34表达数据进行的,其中数千个性状值的测量允许35个强大的推论,同时控制共享的系统发育。在这里,我们提出了一个布朗36运动模型,用于在多种族网络联合37框架下进行定量性状演化,这表明当数千种定量性状平均时,渗入可以产生38个进化的明显收敛模式。我们使用来自野生番茄属40茄的胚珠中的全转录组表达数据来测试我们的理论39预测。检查两个子层都有两者都有特种后渗入的证据,即41,但在其幅度上有很大差异,我们发现进化模式是一致的42,与胚珠基因表达的符号和幅度的渗入历史保持一致。43此外,在渗入速率较高的子层中,我们观察到局部基因树拓扑与表达相似性之间的相关性44,这暗示了渗入45个CIS调节性变化在产生这些宽尺度模式中的作用。48我们的结果揭示了一般作用46在数千种定量性状47的变化模式中渗入的一般作用46,并使用简单的模型信息预测为这些效果提供了一个框架。

Montevallo Today 2023 春夏系列
亲爱的蒙特瓦洛大家庭,当我回顾我们的返校传统时,我想起了今年庆祝活动的主题对于蒙特瓦洛大学来说是多么合适。在许多方面,“传承延续”体现了我们作为一所机构所处的时刻。我们为服务的学生提供方便且负担得起的高等教育机会的传统从未如此重要。我们以多种方式致力于实现这一目标。我很感激,我们的董事会在二月份投票决定连续第六年再次冻结学费。此外,大学每年还为我们的学生提供额外的 100 万美元机构援助。我们感谢许多校友和捐赠者为帮助我们继续这样做而做出的承诺。2023 年,我们计划启动 3000 万美元的募捐活动。该活动将重点关注奖学金。您的礼物已经对我们许多学生的生活产生了巨大影响。其中尤为突出的两位是汉娜·瓦基 (Hannah Waki,2022 届 MBA 毕业生)和阿米莉亚·瓦莱里 (Amelia Valery,2023 届)。汉娜的本科生涯非常辉煌,曾在哈佛大学医学院理查德·卡明斯博士(1974 届)手下实习,今年春天从密歇根大学毕业,获得工商管理硕士学位,将攻读化学博士学位。阿米莉亚获得大众传播学士学位,最近被哥伦比亚大学新闻学院录取。对于每一位捐款的人,我们都非常感谢你们选择以这种方式捐赠奖学金。你们永远不会知道你们的奖学金礼物对我们有多重要,对我们的学生有多重要。我真诚地感谢你们为我们的学生所做的努力。最近,我提醒了他们几个人我最喜欢的一句越南谚语:“吃水果时,要记住种树的人。”对于我们的学生来说,当你们从密歇根大学毕业并在世界上做出伟大的事情时,请记住未来几代的学生也将在这里。

绘制无种子植物的基因组图谱
在陆地定居后的1.5-2亿年左右,陆地植被以无种子植物为主。现代无种子植物是一个并系群落,以苔藓植物(苔类、地钱和角苔)、石松植物和蕨类植物为代表(图1)。从进化角度来看,无种子植物是追溯陆地植物进化重大转变的关键;从应用角度来看,它们是更好地理解种子、果实和花等农学重要性状的生物学的重要外群。无种子谱系的系统发育关系一直存在广泛争议,尤其是苔藓植物之间的关系。几乎所有苔藓、苔类、角苔和维管植物之间的分支顺序的可能组合都是根据形态学、核糖体和/或细胞器DNA证据提出的(参见参考文献1-3)。直到最近,使用转录组和基因组数据集的系统发育基因组学研究才开始提供更明确的答案。Wickett 等人 1 首次应用大量核基因来推断绿色植物的系统发育。在他们的研究中,苔藓和苔类之间的姐妹关系得到了强有力的支持,而角苔的位置则因数据类型(核苷酸与氨基酸)、子集(密码子位置或过滤阈值)和推理方法(连接与物种树方法或最大似然与贝叶斯)1 而异。随后,Puttick 等人 2 和 de Sousa 等人 2 3 使用可以更好地模拟速率和成分异质性的方法重新分析了 Wickett 等人 1 的数据集。这两项研究都证实,苔藓和地钱组成一个进化枝,而 de Sousa 等人 3 则进一步以高置信度将苔藓植物解析为单系植物。然而,应该强调的是,Wickett 等人 1 的数据集中金鱼藻的代表性非常有限,只有两种密切相关的 Nothoceros 物种的转录组。2019 年,随着千株植物 (1KP) 转录组 4 的全面发布,采样更加均衡。1KP 4 和 Harris 等人 5 的分析都支持将金鱼藻置于苔藓和地钱的姐妹地位。最近对金鱼藻基因组的分析进一步支持了所有苔藓植物的单系性 6、7。越来越多的证据表明,现存的陆地植物基本上是由
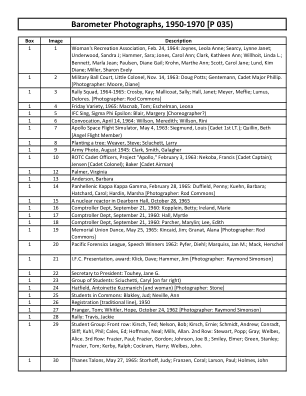
气压计照片,1950-1970 年 [P 035)
盒子图像描述 1 1 妇女娱乐协会,1964 年 2 月 24 日:Joynes, Leola Anne;Searcy, Lynne Janet;Underwood, Sandra J;Hammer, Sara;Jones, Carol Ann;Clark, Kathleen Ann;Willhoit, Linda L.;Bennett, Marla Jean;Paulsen, Diane Gail;Krohn, Marthe Ann;Scott, Carol Jane;Lund, Kim Diane;Miller, Sharon Evely 1 2 军事球场,小上校,1963 年 11 月 14 日:Doug Potts;Gentemann, Phillip 学员少校。[摄影师:Moore, Diane] 1 3 集会小队,1964-1965 年:Crosby, Kay;Mallicoat, Sally;Hall, Janet;Meyer, Meffie;Lumus, Delores。 [摄影师:Rod Commons] 1 4 1965 年星期五综艺节目:Tom Macnab;Leona Eschelman 1 5 IFC Sing,Sigma Phi Epsilon:Margery Blair [编舞?] 1 6 毕业典礼,1964 年 4 月 14 日:Meredith Willson;Rini Willson 1 7 阿波罗太空飞行模拟器,1963 年 5 月 4 日:Louis Siegmund [中尉学员];Beth Quillin [天使飞行成员] 1 8 种树:Steve Weaver;Larry Scluchett 1 9 陆军照片,1945 年 8 月:Smith Clark;Galagher Clark 1 10 ROTC 学员军官,“阿波罗”计划,1963 年 2 月 3 日:Francis Nekoba [上尉学员];贝克 [ 飞行员学员 ] 1 12 帕尔默,弗吉尼亚 1 13 安德森,芭芭拉 1 14 泛希腊 Kappa Kappa Gamma,1965 年 2 月 28 日:达菲尔德,彭妮;库恩,芭芭拉;哈查德,卡罗尔;哈丁,玛莎 [ 摄影师:Rod Commons ] 1 15 迪尔伯恩大厅的核反应堆,1965 年 10 月 28 日 1 16 主计长部,1960 年 9 月 21 日:科普林,贝蒂;爱尔兰,玛丽 1 17 主计长部,1960 年 9 月 21 日:霍尔,默特尔 1 18 主计长部,1960 年 9 月 21 日:帕彻,玛丽琳;李,伊迪丝 1 19 纪念联盟舞会,1965 年 5 月 25 日:金凯德,吉姆; Granat, Alana [摄影师:Rod Commons] 1 20 太平洋法医联盟,1962 年演讲比赛获奖者:Pyfer, Diehl;Marquiss, Jan M.;Mack, Herschel

什么是元基因组学中的binning
宏基因组学是对直接从土壤,水和肠道含量等环境样品中提取的遗传物质的研究,而无需隔离单个生物。该领域使用宏基因组学框来根据相似性将DNA序列分为组。目标是将这些序列分配给其相应的微生物或分类群,从而更深入地了解样本中的微生物多样性和功能。计算方法(例如序列相似性,组成和其他特征)用于分组。宏基因组学的方法包括:基于序列组成的binning,它分析了不同基因组中的不同模式;基于覆盖范围的binning,它使用测序深度将分组读取为垃圾箱;混合式分子,结合了两种方法以提高准确性;基于聚类的封装,可用于高基因组多样性数据集;和基于机器学习的封装,需要带注释的参考基因组进行培训。每种方法都有其优势和局限性,其选择取决于特定的元基因组数据集和研究问题。宏基因组学箱很复杂。2017年,本教程将涵盖元基因组式融合工具,以及咖啡发酵生态系统和metabat 2算法metabat的数据生成MAGS,可以轻松地与下游分析和工具集成,例如分类学注释和功能预测。已经对六个样本进行了测序,生成了6个用于咖啡发酵系统的原始数据集。2。宏基因组套件是分析复杂的微生物群落的关键步骤,但面临着几个挑战,包括水平基因转移污染危险嵌合序列和Maxbin Metabat mycc mycc mycc groopm groopm metawrap anvi'o semibin of de nove bin bin bin bin bin bin bin bin bin bin bin的物种计算工具中的物种计算工具中的应变变化,例如已显示出高度准确的有效扩展和用户友好的基准研究发现,Metabat 2在准确性和计算效率方面都优于其他替代方案,以提供有关宏基因组学软件的更多信息,请参见Sczyrba等。使用Illumina MiSeq全基因组测序进行了六次颞枪i弹枪元基因组研究,以全面分析咖啡微生物组的结构和功能。我们基于这些现实世界数据为本教程创建了模拟数据集。我们将介绍本教程中的以下主题:准备分析历史记录和数据,将metabat 2运行到bin元基因组测序数据。要运行binning,我们首先需要将数据纳入Galaxy,任何分析都应具有自己独特的历史记录。让我们通过单击历史记录面板的顶部创建一个新的历史记录并重命名它。要将序列读取数据上传到星系中,您可以直接从计算机导入它,也可以使用这些链接从Zenodo或数据库中获取它:等等。首先,创建一个名为GTN的文件夹 - 带有主题名称和教程名称的子文件夹的材料。选择所需的文件要从顶部附近的下拉菜单中导入。3。通过在弹出窗口中选择“选择历史记录”,选择要导入数据(或创建新数据)的历史记录。通过重命名示例名称的读取对创建配对集合,然后按照以下步骤:检查所有要包含的数据集,并通过单击“数据集对构建列表”来构建数据集对列表。将未配对的前进和反向读取文本更改为每对的常见选择器。单击“配对这些数据集”以进行有效的前进和反向对。输入一个集合名称,然后单击“创建列表”以构建集合。binning有几个挑战,包括高复杂性,碎片序列,不均匀的覆盖率,不完整或部分基因组,水平基因转移,嵌合序列,应变变异和开放图像1:binning。在本教程中,我们将通过Galaxy使用Metabat 2(Kang等,2019)来学习如何键入元基因组。metabat是“基于丰度和四核苷酸频率的元基因组binning的工具”,该工具将shot弹枪元基因组序列组装到微生物群落中。它使用基因组丰度和四核苷酸频率的经验概率距离来达到98%的精度,并在应变水平下以281个接近完全独特的基因组为准。我们将使用上传的汇编FastA文件作为Metabat的输入,为简单起见保留默认参数。设置为“否”。在输出选项中,“垃圾箱的最小尺寸作为输出”设置为200000。对于ERR2231567样品,有6个箱子,将167个序列分类为第二箱。手:1。4。该工具将在Galaxy版本1.2.9+Galaxy0中使用这些参数:“包含重叠群的Fasta文件”汇编FASTA文件; “考虑融合的良好重叠群的百分比”设置为95; “ binning边缘的最低分数”为60; “每个节点的最大边数”为200; “构建TNF图的TNF概率截止”为0;和“关闭丢失还是小重叠的额外的押金?”The output files generated by MetaBAT 2 include (some are optional and not produced unless required): - Final set of genome bins in FASTA format (.fa) - Summary file with info on each genome bin, including length, completeness, contamination, and taxonomy classification (.txt) - File with mapping results showing contig assignment to a genome bin (.bam) - File containing abundance estimation of each genome bin (.txt) - 每个基因组bin(.txt)的覆盖曲线的文件 - 每个基因组bin的核苷酸组成(.txt) - 文件具有每个基因组bin(.faa)的预测基因序列(.faa)的基因序列,可以进一步分析和用于下游应用,例如功能性注释,相比的植物组合和化学分析,并可以用于下游应用。去复制是识别基因组列表中“相同”的基因组集的过程,并从每个冗余集中删除除“最佳”基因组之外的所有基因组。在重要概念中讨论了相似性阈值以及如何确定最佳基因组。基因组去复制的常见用途是元基因组数据的单个组装,尤其是当从多个样本中组装简短读数时(“共同组装”)。这可能会导致由于组合类似菌株而导致碎片组件。执行共同组装以捕获低丰度微生物。另一种选择是分别组装每个样品,然后去重新复制箱以创建最终的基因组集。metabat 2不会明确执行放松,而是通过利用读取覆盖范围,样品差异覆盖范围和序列组成来提高构架准确性。DREP等工具的设计用于宏基因组学中的复制,旨在保留一组代表性的基因组,以改善下游分析。评估:DREP评估集群中每个基因组的质量,考虑到完整性,污染和应变异质性等因素。基因组选择:在每个群集中,DREP根据用户定义的标准选择代表性基因组。该代表性基因组被认为是群集的“翻译”版本。放松输出:输出包括有关消除基因组的信息,包括身份,完整性和污染。用户可以选择基因组相似性的阈值,以控制删除水平。使用您喜欢的汇编程序分别组装每个样本。bin每个组件分别使用您喜欢的Binner。bin使用您喜欢的Binner共同组装。5。将所有组件中的垃圾箱拉在一起,然后在它们上运行DREP。6。在解复的基因组列表上执行下游分析。检查质量:1。一旦完成,必须检查其质量。2。可以使用CheckM(Parks等,2015)评估binning结果,这是一种用于元基因组学框的软件工具。3。2。检查通过将基因组仓与通用单拷贝标记基因进行比较,评估了基因组仓的完整性和污染。宏基因组学:1。宏基因组学将DNA碎片从混合群落分离为单个垃圾箱,每个垃圾箱代表一个独特的基因组。checkm估计每个基因组箱的完整性(存在的通用单拷贝标记基因集的总数)和污染(在一个以上bin中发现的标记基因的百分比)。关键功能:1。基因组完整性的估计:CheckM使用通用单拷贝标记基因来估计回收基因组的比例。2。基因组污染的估计:CHECKM估计多个箱中存在的标记基因的百分比,表明来自多种生物的潜在DNA。3。识别潜在的杂料:CheckM基于基因组的标记基因分布来识别杂种。4。结果的可视化:CheckM生成图和表,以可视化基因组垃圾箱的完整性,污染和质量指标,从而使解释更加容易。checkm也可以根据与不同分类学组相关的特定标记基因(例如sineage_wf:评估使用谱系特异性标记集对基因组垃圾箱的完整性和污染)进行分类分类的基因组分类。checkm lineage_wf工作流使用标记基因和分类信息的参考数据库来对不同分类学水平的基因组垃圾箱进行分类。来源:-Turaev,D。,&Rattei,T。(2016)。(2014)。使用metabat 2的元基因组重叠群构造教程强调了选择最合适的binning工具的重要性。不同的方法具有不同的优势和局限性,具体取决于所分析的数据类型。通过比较多种封装技术,研究人员可以提高基因组融合的精度和准确性。可用于元基因组数据,包括基于参考的,基于聚类的混合方法和机器学习。每种方法都有其优点和缺点,从而根据研究问题和数据特征使选择过程至关重要。比较多种封装方法的结果有助于确定特定研究的最准确和最可靠的方法。在完整性,污染和应变异质性方面评估所得垃圾箱的质量至关重要。另外,比较已识别基因组的组成和功能谱可以提供有价值的见解。通过仔细选择和比较binning方法,研究人员可以提高基因组箱的质量和可靠性。这最终导致对微生物群落在各种环境中的功能和生态作用有了更好的了解。微生物群落系统生物学的高清晰度:宏基因组学以基因组为中心和应变分辨。- Quince,C.,Walker,A。W.,Simpson,J。T.,Loman,N。J.,&Segata,N。(2017)。shot弹枪宏基因组学,从采样到分析。-Wang,J。和Jia,H。(2016)。元基因组范围的关联研究:微生物组细化。-Kingma,D。P.和Welling,M。(2014年)。自动编码变分贝叶斯。-Nielsen,H。B.等。鉴定和组装基因组和复杂元基因组样品中的遗传因素,而无需使用参考基因组。-Teeling,H.,Meyerdierks,A.,Bauer,M.,Amann,R。,&Glöckner,F。O.(2004)。将四核苷酸频率应用于基因组片段的分配。-Alneberg,J。等。(2014)。通过覆盖范围和组成的结合元基因组重叠群。-Albertsen,M。等。(2013)。通过多个元基因组的差异覆盖层获得的稀有,未培养细菌的基因组序列。-Kang,D.D.,Froula,J.,Egan,R。,&Wang,Z。(2015)。metabat,一种有效的工具,用于准确地重建来自复杂微生物群落的单个基因组。simmons b a和singer s w提出了一种新算法,称为Maxbin 2.0,用于2016年生物信息学期刊中多个元基因组数据集的binning基因组。此外,Kang等人开发了Metabat 2,一种自适应binning算法,该算法于2019年在Peerj发表。PlazaOñate等人引入了MSPMiner,这是一种从shot弹枪元基因组数据重建微生物泛元组的工具,如2019年的生物信息学报道。Other studies like those of Lin and Liao, Chatterji et al, Parks et al, Pasolli et al, Almeida et al, Brooks et al, Sczyrba et al, Qin et al, Bowers et al, Sieber et al, Cleary et al, Huttenhower et al, Saeed et al, and Pride et al have also contributed to the development of metagenomics tools and approaches for genome recovery.这些发现表明,宏基因组分析和计算方法的最新进展使研究人员能够从环境样本中恢复几乎完整的基因组。本文讨论了有关宏基因组学的各种研究,这是对特定环境中多种生物的遗传物质的研究。研究集中于人类肠道微生物组及其在不同人群和年龄之间的组成。引用了几篇论文,其中包括Chen等人的论文。(2020),他开发了一种从宏基因组获得准确而完整的基因组的方法。Daubin等人的另一篇论文。(2003)探讨了细菌基因组中侧向转移基因的来源。本文还提到了有关人肠道微生物组的研究,包括Schloissnig等人的工作。(2013),他绘制了人类肠道微生物组的基因组变异景观。Yatsunenko等。 (2012)研究了在不同年龄和地理位置的人类肠道微生物组。 此外,本文参考了有关微生物从母亲传播到婴儿的研究,包括Asnicar等人的工作。 (2017)和Ferretti等。 (2018)。 本文还涉及宏基因组学分析中使用的机器学习和深度学习技术,例如变化自动编码器和无监督的聚类方法。 最后,本文提到了用于分析元基因组数据的软件工具,包括Li(2013)的BWA-MEM和Paszke等人的Pytorch。 (2019)。 以下是生物信息学和基因组学领域的各种研究文章的摘要。Yatsunenko等。(2012)研究了在不同年龄和地理位置的人类肠道微生物组。此外,本文参考了有关微生物从母亲传播到婴儿的研究,包括Asnicar等人的工作。(2017)和Ferretti等。(2018)。本文还涉及宏基因组学分析中使用的机器学习和深度学习技术,例如变化自动编码器和无监督的聚类方法。最后,本文提到了用于分析元基因组数据的软件工具,包括Li(2013)的BWA-MEM和Paszke等人的Pytorch。(2019)。以下是生物信息学和基因组学领域的各种研究文章的摘要。释义旨在保留原始文章的主要思想和发现,同时以更简洁和易于访问的方式介绍它们。1。**聚类**:一种用于将相似数据点分组在一起的算法,应用于基于Web的数据。2。** art **:用于下一代测序的模拟器可以模仿现实世界数据。3。** metaspades **:一种可以从混合微生物群落中重建基因组的宏基因组组装子。4。** minimap2 **:一种以高精度和速度对齐核苷酸序列的工具。5。** blat **:用于比较基因组序列的爆炸样比对工具。6。** Circos **:用于比较基因组学的可视化工具,用于显示多个基因组之间的关系。7。**高通量ANI分析**:使用平均核苷酸同一性(ANI)指标估算原核基因组之间距离的方法。8。** checkm **:一种评估微生物基因组完整性和污染的工具。9。** BLAST+**:具有改进功能和用户界面的BLAST算法的更新版本。10。** mash **:使用Minhash估算基因组或元基因组距离的工具。11。**浪子**:原核基因组的基因识别和翻译起始位点识别工具。12。** InterPro 2019 **:蛋白质序列注释的InterPro数据库的更新,具有改进的覆盖范围和访问功能。13。14。15。16。**控制虚假发现率**:一种用于管理生物信息学研究中多种假设检验的统计方法。** checkv **:一种用于评估元基因组组装的病毒基因组质量的工具。**使用深度学习从宏基因组数据中识别病毒**:使用机器学习从混合微生物群落中检测病毒的研究。**标准化的细菌分类法**:基于基因组系统发育的细菌进行分类的新框架,该细菌修改了生命之树。17。** gtdb-tk **:一种用于与基因组分类学数据库(GTDB)分类的工具包。18。** iq-Tree **:使用快速有效算法估算最大可能的系统发育的工具。这些摘要概述了生物信息学和基因组学领域的各种研究文章,突出显示了与序列比对,组装,注释和系统发育有关的工具,方法和研究。最新的多个序列对齐软件的进步显着提高了D. M. Mafft版本7,Modelfinder,Astral-III,UFBOOT2,Life V4和APE 5.0等工具的性能和可用性。这些工具通过引入新颖特征,例如快速模型选择,多项式时间种树重建,超快的自举近似和交互式可视化来提高系统发育估计值的准确性。这些软件包的整合已简化了构建进化树的过程,使研究人员可以更轻松地探索复杂的系统发育关系。