XiaoMi-AI文件搜索系统
World File Search System粗粒度

细粒度和粗粒度 SIMD
(i) 细粒度 SIMD:这些实际上是处理实际上由大得多的组件组成的小得多的组件的详细描述。 (ii) 粗粒度 SIMD:这些系统由较少的组件组成,这些组件显然比原始组件多,但比细粒度 SIMD 小得多,但组件的大小比系统的细粒度子组件大得多(高/多)。细粒度和粗粒度 SIMD 架构之间的差异:

粗粒度量子细胞自动机
可以将某些物理演化视为微观离散模型的突发有效结果。受经典粗粒化程序的启发,我们提供了一种遵循 Goldilocks 规则的粗粒化色盲量子细胞自动机的简单程序。该程序包括 (i) 将量子细胞自动机 (QCA) 在时空上分组为大小为 N 的细胞;(ii) 将细胞的状态投射到其边界上,并将其与精细动力学联系起来;(iii) 通过边界状态描述整体动力学,我们称之为信号;(iv) 为不同大小为 N 的细胞构建粗粒化动力学。这个简单的玩具模型的副产品是斯托克斯定律的一般离散模拟。此外,我们证明在时空极限中,自动机收敛到狄拉克自由哈密顿量。我们在这里介绍的 QCA 可以通过当今的量子平台实现,例如里德堡阵列、捕获离子和超导量子比特。我们希望我们的研究能够为更深入地理解这些分辨率有限的系统铺平道路。

细化粗粒度分子拓扑
摘要 分子动力学 (MD) 模拟对于预测不同分子体系的物理和化学性质至关重要。虽然全原子 (AA) MD 提供了高精度,但其计算成本高昂,这促使了粗粒度 MD (CGMD) 的发展。CGMD 将分子结构简化为具有代表性的微珠,以降低成本,但会牺牲精度。像 Martini3 这样的 CGMD 方法,经过实验数据校准后,在各个分子类别中具有良好的泛化能力,但往往无法满足特定领域应用的精度要求。本研究引入了一种基于贝叶斯优化的方法来优化 Martini3 拓扑结构,使其能够适应特定应用,从而确保精度和效率。优化后的 CG 势能适用于任何聚合度,提供与 AA 模拟相当的精度,同时保持与 CGMD 相当的计算速度。通过弥合效率和精度之间的差距,该方法推动了多尺度分子模拟的发展,使各个科学技术领域能够以经济高效的方式发现分子。 1. 引言粗粒度分子动力学 (CGMD) 1,2 已成为材料开发的重要工具,为了解聚合物 3 、蛋白质 4 和膜 5 等复杂分子系统提供了关键信息。CGMD 的主要优势在于它能够在更大长度尺度和更长时间范围内探索分子现象,超越了传统全原子分子动力学 (AAMD) 6–8 模拟的能力,后者通常提供更高的分辨率,因此特别擅长捕捉详细的界面相互作用 9 。具体而言,CGMD 通过将原子团有效地表示为珠子 10–15 来实现这种加速,从而将模拟能力在时间上从皮秒扩展到微秒,在空间上从纳米扩展到微米。因此,粗粒度技术为传统 AAMD 无法获得的复杂分子现象提供了前所未有的洞察,从而能够研究聚合物自组装行为等复杂现象 16 。新兴的CGMD建模工具集依赖于两个关键组件来学习潜在的分子间关系:珠子映射方案和珠子间相互作用的参数化。这些组件的开发主要采用两种方法:自上而下10–12和自下而上13–

马提尼 3 粗粒度力场:小分子
Martini 粗粒度力场 Martini 3 的最新重新参数化提高了该模型在预测分子动力学模拟中的分子堆积和相互作用方面的准确性。在这里,我们描述了如何在 Martini 3 框架内精确参数化小分子,并提供了一个经过验证的小分子模型数据库。我们特别关注脂肪族和芳香族环状结构的描述,这些结构在溶剂和药物等小分子或蛋白质和合成聚合物等大分子的构成块中普遍存在。在 Martini 3 中,环状结构由使用更高分辨率粗粒度颗粒(小颗粒和微小颗粒)的模型描述。因此,本数据库构成了校准新 Martini 3 小颗粒和微小颗粒尺寸的基石之一。这些模型表现出出色的分配行为和溶剂性能。还捕获了不同本体相之间的可混溶性趋势,从而完成了参数化过程中考虑的一组热力学性质。我们还展示了新的珠子尺寸如何能够很好地表示分子体积,从而转化为更好的结构特性,例如堆叠距离。我们进一步介绍了设计策略,以构建复杂度更高的小分子的 Martini 3 模型。

可转移、可极化的蛋白质粗粒度模型─ProMPT
摘要:由于表示所有原子的计算复杂性,经典分子动力学 (MD) 模拟在原子分辨率(细粒度级别,FG)下对大多数生物分子过程的应用仍然有限。这个问题在具有非常大构象空间的基于蛋白质的生物分子系统存在的情况下被放大,并且具有细粒度分辨率的 MD 模拟具有探索该空间的缓慢动态。文献中当前的可转移粗粒度 (CG) 力场要么仅限于以隐式形式编码环境的肽,要么无法捕获从氨基酸一级序列到二级/三级肽结构的转变。在这项工作中,我们提出了一种可转移的 CG 力场,它明确表示环境,以便对蛋白质进行精确模拟。力场由一组代表不同化学基团的伪原子组成,这些化学基团可以连接/关联在一起以创建不同的生物分子系统。这保留了力场在多种环境和模拟条件中的可转移性。我们添加了可以响应环境异质性/波动的电子极化,并将其与蛋白质的结构转变耦合。非键合相互作用通过基于物理的特征(例如通过热力学计算确定的溶剂化和分配自由能)进行参数化,并与实验和/或原子模拟相匹配。键合势是从非冗余蛋白质结构数据库中的相应分布推断出来的。我们通过模拟经过充分研究的水蛋白系统来验证 CG 模型,这些系统具有特定的蛋白质折叠类型 Trp-cage、Trpzip4、villin、WW-domain 和 β - α - β 。我们还探索了力场在研究 A β 16-22 肽的水聚集中的应用。■ 简介蛋白质分子的生理功能与其相关结构和动力学密切相关。1、2
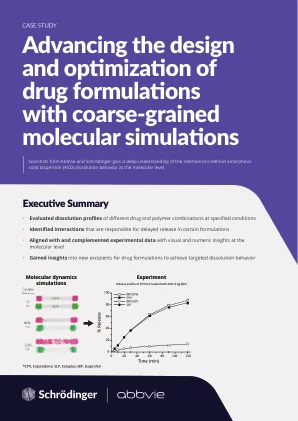
案例研究利用粗粒度分子模拟推进药物配方的设计和优化
在亲水性聚合物基质中配制低水溶性小分子药物,也称为无定形固体分散体 (ASD),是实现有效药物输送和生物利用度的最常见方法之一。生产高性能 ASD 取决于各种因素,例如药物赋形剂基质的物理稳定性、其在溶解过程中与聚合物的相互作用以及药物在水性介质中的释放速率。通常,研究人员会进行大量的设计和实验迭代来实现这一目标。虽然可以从实验数据中得出关于药物释放行为的假设,但对基本机制的全面理解和对分子水平事件的洞察仍然难以实现。仅通过实验很难获得详细的药物/聚合物/水相互作用。因此,需要一种更有效的方法来指导为特定药物选择合适的赋形剂(包括聚合物)。

通过粗粒
蛋白XPA在核苷酸切除修复途径中起关键作用。最近的实验工作表明,XPA的功能动力学涉及沿DNA的一维扩散以搜索损伤位点。在这里,我们使用各种盐浓度的广泛的粗粒分子模拟来研究所涉及的动力学过程。结果表明扩散机制的盐浓度依赖性很强。在低盐浓度下,与旋转耦合的一维扩散是主要机制。在高盐浓度下,三维机制的扩散变得更有可能。在较广泛的盐浓度下,涉及DNA结合的残基是相似的,并且沿DNA显示的XPA的一维扩散是降低功能。此亚延伸功能暂定归因于XPA – DNA相互作用的各种强度。另外,我们表明,与DNA的结合和盐浓度升高倾向于拉伸XPA的构象,从而增加了位点的暴露范围,以结合其他修复蛋白。
