XiaoMi-AI文件搜索系统
World File Search System系统学

分子系统和系统发育分析,对氢氯巴鲁斯Sp的共生细菌。产生抗生素酶藻酸盐裂解酶
摘要。藻类细菌群落以生产破坏藻酸盐的抗生素酶而闻名,这些酶是生物膜的主要成分的藻酸盐。生物膜相关感染是危险的,因为它们对抗生素和人类免疫系统产生了抗性。这项工作报告了基于分子系统学和系统发育分析16S rRNA的几种海洋藻素细菌,可能是新的物种。它们是从不同的棕色藻类氢层sp中分离出来的。居住在印度尼西亚Wakatobi的Hoga岛周围的海洋中。这项研究旨在揭示这些细菌分离株的分子身份和亲属关系,以理解其更多的特性,即氢氯拉斯sp的共生体。分子鉴定和系统发育树的结构是根据使用27F-1492R引物的聚合酶链反应对16S rRNA基因扩增的序列进行的。可以获得总共31种棕色藻类氢氯拉鲁斯共生细菌的分离株,表明藻类是海洋细菌的有吸引力的共生菌宿主。能够产生藻酸盐裂解酶和琼脂酶的分离株数量为15。然而,在用最小藻酸盐培养基进行确认测试后,只有15个分离株中只有12个是藻酸盐裂解酶生产者。在具有最高藻体级指数的8个分离物上的分子鉴定显示了与3种不同属的最接近的关系:颤音,拟南芥和aestuariibacter。基于BLAST(基本局部对齐搜索工具)分析,5比其对齐结果的最高命中率低于97%的相似性水平,表明它们可能是新物种。这些发现表明了海洋棕色藻类氢层sp的潜力。是藻素溶液的潜在宿主。关键词:琼脂酶,藻酸盐裂解酶,海洋细菌,瓦卡托比。简介。抗生素酶是可用于控制和去除细菌生物膜的酶的类型。这些酶溶解了包含细菌细胞外基质的多糖,蛋白质和核酸。抗生素酶包括脂肪酶,可防止纤维旁溶血生物膜和纤维素酶的生长,这些脂肪酶会分解大多数生物膜中存在的纤维素(Gutiérrez2019)。也已经证明了脂肪酶,纤维酶和蛋白酶K等组合酶在预防和消除副溶血性生物膜上有效(Li et al 2022)。其他生物膜控制酶包括β-葡萄糖酶,蛋白酶和淀粉酶,它们可以分解EPS基质并防止生物膜的产生。抗生素酶被认为比传统方法更有效,更环保,例如侵袭性化学物质,例如氢氧化钠或次氯酸钠,它们可以腐蚀机械和材料(Blackman 2021)。

使用...
摘要:随着电子商务和在线交易的快速扩展,在线支付欺诈检测,付款欺诈的风险已成为企业和消费者的重大关注点。本项目着重于在线支付欺诈检测系统的开发和实施,利用高级机器学习算法和数据分析技术。通过分析交易数据,用户行为模式和上下文信息,该系统旨在实时识别和防止欺诈活动。通过数据预处理,功能工程和模型培训,该系统学会了区分合法和欺诈性交易。使用各种机器学习算法,包括逻辑回归,随机森林和神经网络,用于检测指示欺诈行为的异常和模式。评估指标(例如精确度,召回和F1得分)用于评估系统的性能,并确保其在检测欺诈交易方面的有效性,同时最大程度地减少误报。此外,该项目还探讨了实时数据流,异常检测技术和行为生物识别技术的集成,以进一步增强系统的欺诈检测能力。最终,开发的在线支付欺诈检测系统是保护企业和消费者免受财务损失并保留对在线支付生态系统的信任的关键工具。但是,这种扩展也导致了在线支付欺诈的增加,对数字支付系统的安全性和可信度构成了重大挑战。关键字:在线支付欺诈,机器学习,欺诈检测,电子商务安全,异常检测,数据分析时间监测,行为生物识别技术I引言电子商务和在线交易的快速增长为消费者和企业带来了前所未有的便利性。欺诈活动可能会导致重大财务损失,对品牌声誉的损失以及消费者信任的下降。因此,开发强大的欺诈检测机制对于保护其运营和客户群的企业至关重要。此项目旨在使用高级机器学习技术和数据分析设计和实施在线付款欺诈检测系统。通过分析大量的交易数据和用户行为模式,系统试图实时识别和防止欺诈性交易。检测过程涉及数据预处理,功能工程以及各种机器学习算法的应用,例如逻辑回归,随机森林和神经网络。这些模型经过训练,以识别指示欺诈的模式和异常,从而实现主动措施。除了传统的机器学习方法外,该项目探讨了实时数据流和行为生物识别技术的整合以提高欺诈检测的准确性和效率。使用精度,召回和F1得分等指标评估系统的性能,从而确保检测欺诈交易和最小化假阳性之间的平衡。通过提供全面且可扩展的解决方案,该项目有助于保护企业和消费者免受在线支付欺诈的侵害,最终促进了更安全的

从环境DNA metabarcoding推断功能多样性
Aglieri,G.,Baillie,C.,Mariani,S.,Cattano,C.,Calò,A.,Turco,G.,Spatafora,D.,Di Franco,A.环境DNA有效地捕获了沿海鱼类社区的功能多样性。分子生态学,30(13),3127–3139。https://doi.org/10.1111/mec.15661 Albert,J。S.,Destouni,G.,Duke-Sylvester,S.M.,Magurran,A.E.(2021)。科学家对淡水生物多样性危机的人类警告。Ambio,50,85–94。https://doi.org/10.1007/s1328 0-020-01318 -8 Albert,J.S.,Tagliacollo,V.A。,&Dagosta,F。(2020)。新热带淡水鱼的多样化。生态,进化和系统学的年度审查,51(1),27-53。https://doi.org/10.1146/annur ev- ecols YS-01162 0-031032 BELLEMAIN,E.,CARLSEN,T.,BROCHMANN,C.,COISSAC,COISSAC,E.它是真菌的环境DNA条形码:一种硅方法揭示了潜在的PCR偏见。BMC微生物学,10(189),189。https:// doi。org/10.1186/1471-2180-10-189 Boyer,F.,Mercier,C.,Bonin,A.,Le Bras,Y.,Taberlet,P。,&Coissac,E。(2016年)。obitools:用于DNA ME- TABARCODING的UNIX启发的软件包。分子生态资源,16(1),176–182。https://doi.org/10.1111/1755-0998.12428 Brosse,S.,Charpin,N.,Guohuan,S.,Toussaint,A.A.,Tedesco,P。和Villeger,S。(2021)。Fishmorph:淡水鱼形态特征的全球数据库。(2021)。全球生态和生物地理学,30,2330–2336。https://doi.org/10.1111/GEB.13395 Cantera,I.,Cilleros,K.,Valentini,A.,A.,A.,Dejean,A.,Dejean,T.,Iribar,A. 为热带流和河流中的鱼类库存优化环境DNA采样工作。 科学报告,9(1),3085 M.,Salguero-Gómez,R.,Vásquez-Valderrama,M。和Toussaint,A。 遍布整个生命树的全球功能多样性。 科学进步,7(13),EABF2675。 https://doi.org/10.1126/sciadv.abf2675 Cilleros,K.,Valentini,A. 使用环境DNA(EDNA)在高分化环境中解锁生物多样性和保护研究:圭亚那的测试https://doi.org/10.1111/GEB.13395 Cantera,I.,Cilleros,K.,Valentini,A.,A.,A.,Dejean,A.,Dejean,T.,Iribar,A.为热带流和河流中的鱼类库存优化环境DNA采样工作。科学报告,9(1),3085 M.,Salguero-Gómez,R.,Vásquez-Valderrama,M。和Toussaint,A。遍布整个生命树的全球功能多样性。科学进步,7(13),EABF2675。https://doi.org/10.1126/sciadv.abf2675 Cilleros,K.,Valentini,A.使用环境DNA(EDNA)在高分化环境中解锁生物多样性和保护研究:圭亚那的测试

GU/ACAD - PG/BOS -NEP/2024/251 DATE 果阿大学 PG/BOS -NEP/2024-25/780日期:24.01.2025圆形ref risea- 2025 GU/Acad-Admission/Ph.D./10月/2024/63日期:25/... 生活世界:什么是生活?生物多样性;需要分类;生命的三个领域;分类学和系统学;物种和分类群的概念 Infofest GU/ACAD - PG/BOS -NEP ENGG./2024-25/770日期:22.01。 ... 入学排名测试(GU-ART)2025-26
A candidate for being eligible for admission to the Diploma in Clinical Genetics and Medical Laboratory Techniques must have passed the examination of degree of Bachelor of Science of this University or from any other recognized University, with at least 32 credits in the first, second and third years taken together under the Choice Based Credit System (CBCS) in the subject of either Zoology, Microbiology, Biotechnology, Chemistry/Biochemistry.在学期的考试系统中,论文或单位的数量应为14,总数为1400分,其中八篇论文是F.Y.的研究。和S.Y.T.Y.一起审算至少6篇论文。有资格参加该主题的PG入学。

微生物取证和计算生物学
[19] Kunin,V.,Copeland,A.,Lapidus,A.,Mavromatis,K。,&Hugenholtz,P。(2008)。宏基因组学的生物信息学指南。微生物学和分子生物学评论,72(4),557-578。[20] Jolley,K。A.,Chan,M。S.,&Maiden,M.C。(2004)。MLSTDBNET分布的多洛克斯序列键入(MLST)数据库。BMC生物信息学,5(1),86。[21] Enright,M。C.和Spratt,B。G.(1999)。多焦点序列键入。微生物学的趋势,7(12),482-487。[22] Healy,M.,Huong,J.,Bittner,T.,Lising,M.,Frye,S.,Raza,S。,&Woods,C。(2005)。通过自动重复序列的PCR键入微生物DNA。临床微生物学杂志,第43(1)期,199-207。[23] Vergnaud,G。和Pourcel,C。(2006)。多个基因座VNTR(串联重复的可变数量)分析。分子鉴定,系统学和原核生物的种群结构,83-104。[24] Van Belkum,A。(2007)。通过多焦点数量的串联重复分析(MLVA)来追踪细菌物种的分离株。病原体和疾病,49(1),22-27。[25] Vergnaud,G。和Pourcel,C。(2009)。多个基因座变量串联重复分析数。微生物的分子流行病学:方法和方案,141-158。[26] Fricke,W。F.,Rasko,D。A.和Ravel,J。(2009)。基因组学在鉴定,预测和预防生物学威胁中的作用。PLOS Biology,7(10),E1000217。[27] Wu,M。和Eisen,J。A.(2008)。95-100)。一种简单,快速且准确的系统基因推断方法。基因组生物学,9(10),R151。[28] Liu,B.,Gibbons,T.,Ghodsi,M。和Pop,M。(2010年12月)。隐式:元基因组序列的分类分析。生物信息学和生物医学(BIBM),2010年IEEE国际会议(pp。IEEE。 [29] Wang,Z。,&Wu,M。(2013)。 门水平细菌系统发育标记数据库。 分子生物学与进化,30(6),1258-1262。 [30] Darling,A。E.,Jospin,G.,Lowe,E.,Matsen IV,F。A.,Bik,H。M.,&Eisen,J. A. (2014)。 系统缩影:基因组和宏基因组的系统发育分析。 peerj,2,e243。 [31] Taberlet,P.,Prud'Homme,S.M.,Campione,E.,Roy,J.,Miquel,C.,Shehzad,W。,&Melodelima,C。(2012)。 土壤采样和细胞外DNA的分离,适用于大量的起始材料。 分子生态学,21(8),1816-1820。IEEE。[29] Wang,Z。,&Wu,M。(2013)。门水平细菌系统发育标记数据库。分子生物学与进化,30(6),1258-1262。[30] Darling,A。E.,Jospin,G.,Lowe,E.,Matsen IV,F。A.,Bik,H。M.,&Eisen,J.A.(2014)。系统缩影:基因组和宏基因组的系统发育分析。peerj,2,e243。[31] Taberlet,P.,Prud'Homme,S.M.,Campione,E.,Roy,J.,Miquel,C.,Shehzad,W。,&Melodelima,C。(2012)。土壤采样和细胞外DNA的分离,适用于大量的起始材料。分子生态学,21(8),1816-1820。

海岸鱼零售的DNA条形码参考库
Avise,J。C.(1989)。分子标记,自然历史和进化。纽约,纽约:施普林格。Bellwood,D。R.和Meyer,C。P.(2009)。 在海洋生物多样性热点中寻找热量。 生物地理学杂志,36,569–576。 https://doi.org/10.1111/j.1365-2699.2008.02029.x Bermingham,E.,McCafferty,S。,&Martin,A。P.(1997)。 鱼类生物地球和分子时钟:来自巴拿马伊斯兰教的观点。 在T. D. Kocher和C. A. Stepien(编辑) ),鱼类的分子系统学(pp。 113–128)。 圣地亚哥,加利福尼亚州:学术出版社。 Bouckaert,R。R.,Heled,J.,Kühnert,D.,Vaughan,T.,Wu,C.-H.,Xie,D. (2014)。 野兽2:用于贝叶斯进化分析的软件平台。 PLOS计算生物学,10,E1003537。 https://doi.org/10.1371/journ al.pcbi.1003537 Chase,J.M。,&Leibold,M。A. (2002)。 空间量表决定了生产力的关系。 自然,416,427–430。 https:// doi。 org/10.1038/416427a Chin,T。C.,Adibah,A。 B.,Danial Hariz,Z。 A.和Siti Azizah,M。N.(2016)。 通过DNA钢筋编码:提高食品市场的透明度来检测马来西亚标记错误的海鲜产品。 食品控制,64,247–256。 https://doi.org/10.1016/j.foodc ont.2015.11.042 Cinner,J.E.,Graham,N.A。J. (2013)。 在珊瑚礁渔业状况下,当地人口密度和与市场距离的全球影响。 罗马。Bellwood,D。R.和Meyer,C。P.(2009)。在海洋生物多样性热点中寻找热量。生物地理学杂志,36,569–576。https://doi.org/10.1111/j.1365-2699.2008.02029.x Bermingham,E.,McCafferty,S。,&Martin,A。P.(1997)。鱼类生物地球和分子时钟:来自巴拿马伊斯兰教的观点。在T. D. Kocher和C. A. Stepien(编辑),鱼类的分子系统学(pp。113–128)。圣地亚哥,加利福尼亚州:学术出版社。 Bouckaert,R。R.,Heled,J.,Kühnert,D.,Vaughan,T.,Wu,C.-H.,Xie,D. (2014)。 野兽2:用于贝叶斯进化分析的软件平台。 PLOS计算生物学,10,E1003537。 https://doi.org/10.1371/journ al.pcbi.1003537 Chase,J.M。,&Leibold,M。A. (2002)。 空间量表决定了生产力的关系。 自然,416,427–430。 https:// doi。 org/10.1038/416427a Chin,T。C.,Adibah,A。 B.,Danial Hariz,Z。 A.和Siti Azizah,M。N.(2016)。 通过DNA钢筋编码:提高食品市场的透明度来检测马来西亚标记错误的海鲜产品。 食品控制,64,247–256。 https://doi.org/10.1016/j.foodc ont.2015.11.042 Cinner,J.E.,Graham,N.A。J. (2013)。 在珊瑚礁渔业状况下,当地人口密度和与市场距离的全球影响。 罗马。圣地亚哥,加利福尼亚州:学术出版社。Bouckaert,R。R.,Heled,J.,Kühnert,D.,Vaughan,T.,Wu,C.-H.,Xie,D.(2014)。野兽2:用于贝叶斯进化分析的软件平台。PLOS计算生物学,10,E1003537。https://doi.org/10.1371/journ al.pcbi.1003537 Chase,J.M。,&Leibold,M。A.(2002)。空间量表决定了生产力的关系。自然,416,427–430。https:// doi。org/10.1038/416427a Chin,T。C.,Adibah,A。B.,Danial Hariz,Z。 A.和Siti Azizah,M。N.(2016)。 通过DNA钢筋编码:提高食品市场的透明度来检测马来西亚标记错误的海鲜产品。 食品控制,64,247–256。 https://doi.org/10.1016/j.foodc ont.2015.11.042 Cinner,J.E.,Graham,N.A。J. (2013)。 在珊瑚礁渔业状况下,当地人口密度和与市场距离的全球影响。 罗马。B.,Danial Hariz,Z。A.和Siti Azizah,M。N.(2016)。通过DNA钢筋编码:提高食品市场的透明度来检测马来西亚标记错误的海鲜产品。食品控制,64,247–256。https://doi.org/10.1016/j.foodc ont.2015.11.042 Cinner,J.E.,Graham,N.A。J.(2013)。在珊瑚礁渔业状况下,当地人口密度和与市场距离的全球影响。罗马。保护生物学,27,453–458。https://doi.org/10.1111/j.1523-1739.2012.01933.x Cinner,J.E.,Huchery,C.,Macneil,M.A.,Graham,N.A.J.世界珊瑚礁之间的亮点。自然,535(7612),416–419。https://doi.org/10.1038/ Natur E18607 Collet,A.,Durand,J.D.,Desmarais,E.DNA条形码在大型后可以改善有关鱼类生物多样性的知识:来自SW印度洋La Reunion的一个例子。线粒体DNA A部分,29(6),905–918。https:// doi。org/10.1080/24701 394.2017.1383406 Collins,R.A。,&Cruickshank,R.H。(2014)。已知已知的未知数未知数未知和未知数在DNA Barcoding中:对Dowton等人系统生物学的评论,63(6),1005–1009。https://doi.org/10.1093/sysy.1093/sysysbi O/syu060 delrieu-delrieu--trottin,E.,e.,j。t。法国波利尼西亚海岸鱼类的DNA条形码参考库。科学数据,6(1),114。https:// doi。org/10.1038/s4159 7-019-0123-5 Di Pinto,A.,Marchetti,P.,Mottola,A.,Bozzo,G.,G.,Bonerba,E.使用DNA条形码在鱼片中的物种鉴定。渔业研究,170,9-13。https:// doi。org/10.1016/j.fishr es.2015.05.006 Durand,J.-D.,Hubert,N.,Shen,K.-N。,&Borsa,P。(2017)。DNA灰色mul虫。《鱼类生物学与渔业》中的评论,27(1),233-243。粮农组织(2018)。https://doi.org/10.1007/s1116 0-016-9457-7 Erdmann,M。,&Allen,G。R.(2012)。 东印度群岛的礁鱼。 珀斯,澳大利亚:嗯。 la Cheation MondialedesPêches等人2018。 atteindre les objectifs dedévelopment耐用。 Ficetola,G。F.,Miaud,C.,Pompanon,F。,&Taberlet,P。(2008)。 使用水样中的环境DNA检测物种。 生物学信,4,423–425。 https://doi.org/10.1098/rsbl.2008.0118 Froese,R。,&Pauly,D。(2014)。 fishbase .http://www.fishb ase.org,electronic版本访问了11/2019。 Fujisawa,T。和Barraclough,T。G.(2013)。 使用Single-Locus数据和广义混合Yule Colescent方法对物种进行分解:对模拟数据集的修订方法和评估。 系统生物学,62(5),707–724。 https://doi.org/10.1093/sysbi o/syt033 Gaboriau,T.,Leprieur,F.,Mouillot,D。,&Hubert,N。(2018)。 物种地理位置对印度太平洋地区珊瑚礁鱼类生物多样性当前模式的影响。 Ecograph,40,1295–1306。 https://doi.org/10.1111/ecog.02589https://doi.org/10.1007/s1116 0-016-9457-7 Erdmann,M。,&Allen,G。R.(2012)。东印度群岛的礁鱼。珀斯,澳大利亚:嗯。la Cheation MondialedesPêches等人2018。atteindre les objectifs dedévelopment耐用。Ficetola,G。F.,Miaud,C.,Pompanon,F。,&Taberlet,P。(2008)。使用水样中的环境DNA检测物种。生物学信,4,423–425。https://doi.org/10.1098/rsbl.2008.0118 Froese,R。,&Pauly,D。(2014)。fishbase .http://www.fishb ase.org,electronic版本访问了11/2019。Fujisawa,T。和Barraclough,T。G.(2013)。使用Single-Locus数据和广义混合Yule Colescent方法对物种进行分解:对模拟数据集的修订方法和评估。系统生物学,62(5),707–724。https://doi.org/10.1093/sysbi o/syt033 Gaboriau,T.,Leprieur,F.,Mouillot,D。,&Hubert,N。(2018)。物种地理位置对印度太平洋地区珊瑚礁鱼类生物多样性当前模式的影响。Ecograph,40,1295–1306。https://doi.org/10.1111/ecog.02589

GU/ACAD - PG/BOS -NEP/2024/2024/151 生物多样性I(微生物,藻类,真菌和苔藓植物) 课程提纲,用于溪流变化 /生物化学中的gu-art < / div> GU/ACAD - PG/BOS -NEP/2024/251 DATE 果阿大学 PG/BOS -NEP/2024-25/780日期:24.01.2025圆形ref risea- 2025 GU/Acad-Admission/Ph.D./10月/2024/63日期:25/... 生活世界:什么是生活?生物多样性;需要分类;生命的三个领域;分类学和系统学;物种和分类群的概念 Infofest GU/ACAD - PG/BOS -NEP ENGG./2024-25/770日期:22.01。 ... 入学排名测试(GU-ART)2025-26
心理学实践实验:1。学习有意义且毫无意义的言语材料的功效2。召回和识别作为保留测试的比较研究3。注意力的波动4。光学错觉:Muller - Lyer 5。深度感知6。反应时间7。stroop效果8。概念形成(使用卡片或块)9。Zeigarnik效应
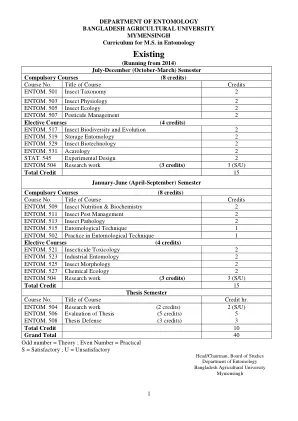
昆虫学系
动物作品,现场指南和手册,手册,目录,清单,新分类单元的描述,进化和生物出版物。推荐书籍:教科书Mayr,E。1960。系统动物学原理。McGraw-Hill Book Co.,纽约。Mayr,E。Linsley,例如 和使用者R.L. 1953。 系统动物学的方法和原理。 McGraw-Hill Book Co.,纽约。 Sokal,R.R。 和Sneath,P.H.A。 1963。 数值分类法的原则,W.H。 Freeman and Co.旧金山。 参考书Abivardi,C。1999。 伊拉伊昆虫学。 卷。 1,荷兰施普林格。 Bisby,F.A。 Vaughan,J.G。 和Wright,C.A。 1980。 Chaemosystematics,原理和实践。 学术出版社,纽约。 Chamberlin,W.J。 1952。 昆虫学命名法和文学。 wn。 C. Brown,爱荷华州迪比克。 Goto,H.E。 1982。 动物分类法,爱德华·阿诺德,伦敦。 Kapoor,V.C。 1990。 昆虫的起源和进化。 publ。 Kallayani,新德里。 Kim,E.E.C。 和McPheron,B.A。 1993。 害虫patterns和变异的进化。 Intercept Ltd.,伦敦。 Leone,C.A。 1964。 分类生物化学和血清学。 Ronald Press,堪萨斯州。 Lincoln,R.J。 Boxshall,G.A。 和Clark,P.F。 1981。Mayr,E。Linsley,例如和使用者R.L.1953。系统动物学的方法和原理。McGraw-Hill Book Co.,纽约。Sokal,R.R。和Sneath,P.H.A。1963。数值分类法的原则,W.H。Freeman and Co.旧金山。参考书Abivardi,C。1999。伊拉伊昆虫学。卷。1,荷兰施普林格。Bisby,F.A。Vaughan,J.G。 和Wright,C.A。 1980。 Chaemosystematics,原理和实践。 学术出版社,纽约。 Chamberlin,W.J。 1952。 昆虫学命名法和文学。 wn。 C. Brown,爱荷华州迪比克。 Goto,H.E。 1982。 动物分类法,爱德华·阿诺德,伦敦。 Kapoor,V.C。 1990。 昆虫的起源和进化。 publ。 Kallayani,新德里。 Kim,E.E.C。 和McPheron,B.A。 1993。 害虫patterns和变异的进化。 Intercept Ltd.,伦敦。 Leone,C.A。 1964。 分类生物化学和血清学。 Ronald Press,堪萨斯州。 Lincoln,R.J。 Boxshall,G.A。 和Clark,P.F。 1981。Vaughan,J.G。和Wright,C.A。1980。Chaemosystematics,原理和实践。学术出版社,纽约。Chamberlin,W.J。1952。昆虫学命名法和文学。wn。C. Brown,爱荷华州迪比克。 Goto,H.E。 1982。 动物分类法,爱德华·阿诺德,伦敦。 Kapoor,V.C。 1990。 昆虫的起源和进化。 publ。 Kallayani,新德里。 Kim,E.E.C。 和McPheron,B.A。 1993。 害虫patterns和变异的进化。 Intercept Ltd.,伦敦。 Leone,C.A。 1964。 分类生物化学和血清学。 Ronald Press,堪萨斯州。 Lincoln,R.J。 Boxshall,G.A。 和Clark,P.F。 1981。C. Brown,爱荷华州迪比克。Goto,H.E。 1982。 动物分类法,爱德华·阿诺德,伦敦。 Kapoor,V.C。 1990。 昆虫的起源和进化。 publ。 Kallayani,新德里。 Kim,E.E.C。 和McPheron,B.A。 1993。 害虫patterns和变异的进化。 Intercept Ltd.,伦敦。 Leone,C.A。 1964。 分类生物化学和血清学。 Ronald Press,堪萨斯州。 Lincoln,R.J。 Boxshall,G.A。 和Clark,P.F。 1981。Goto,H.E。1982。动物分类法,爱德华·阿诺德,伦敦。Kapoor,V.C。 1990。 昆虫的起源和进化。 publ。 Kallayani,新德里。 Kim,E.E.C。 和McPheron,B.A。 1993。 害虫patterns和变异的进化。 Intercept Ltd.,伦敦。 Leone,C.A。 1964。 分类生物化学和血清学。 Ronald Press,堪萨斯州。 Lincoln,R.J。 Boxshall,G.A。 和Clark,P.F。 1981。Kapoor,V.C。1990。昆虫的起源和进化。publ。Kallayani,新德里。 Kim,E.E.C。 和McPheron,B.A。 1993。 害虫patterns和变异的进化。 Intercept Ltd.,伦敦。 Leone,C.A。 1964。 分类生物化学和血清学。 Ronald Press,堪萨斯州。 Lincoln,R.J。 Boxshall,G.A。 和Clark,P.F。 1981。Kallayani,新德里。Kim,E.E.C。 和McPheron,B.A。 1993。 害虫patterns和变异的进化。 Intercept Ltd.,伦敦。 Leone,C.A。 1964。 分类生物化学和血清学。 Ronald Press,堪萨斯州。 Lincoln,R.J。 Boxshall,G.A。 和Clark,P.F。 1981。Kim,E.E.C。和McPheron,B.A。1993。害虫patterns和变异的进化。Intercept Ltd.,伦敦。Leone,C.A。 1964。 分类生物化学和血清学。 Ronald Press,堪萨斯州。 Lincoln,R.J。 Boxshall,G.A。 和Clark,P.F。 1981。Leone,C.A。1964。分类生物化学和血清学。Ronald Press,堪萨斯州。 Lincoln,R.J。 Boxshall,G.A。 和Clark,P.F。 1981。Ronald Press,堪萨斯州。Lincoln,R.J。 Boxshall,G.A。 和Clark,P.F。 1981。Lincoln,R.J。 Boxshall,G.A。和Clark,P.F。1981。生态,进化和系统学的字典。剑桥大学。按,剑桥。

微生物学中的属是什么
nlm提供了对科学文献的访问,而无需暗示与内容的认可或一致。分类法涉及根据特征对微生物进行分类,细菌通过革兰氏染色反应分为两个主要组,并表现出各种形状和大小。在临床实践中,细菌是通过形态学,氧的需求和生化测试对细菌进行分类的。基因探针和基于PCR的技术等诊断测试系统检测特定细菌。细菌物种通常根据基因重组频率表现出不同的种群结构。键入分离株对于流行病学研究和监视至关重要。微生物可以分为七个大型生物群:藻类,原生动物,粘液霉菌,真菌,细菌,古细菌和病毒。藻类,原生动物,粘液霉菌和真菌是真核微生物,具有类似于动植物的细胞结构。细菌,包括支原体,立克群和衣原体组,具有原核组织。古细菌是一群独特的原核生物,与其他生物没有密切的祖先关系。只有细菌和病毒在医学或兽医上被认为是重要的。病毒是具有简单结构和不同繁殖模式的最小传染剂。病毒,无蛋白质的RNA片段,引起植物的疾病,而prion是动物和人类致命神经退行性疾病的病因。传染性同工型中发生构成变化(第60章)。系统学也称为系统发育学。分类法包括三个组成部分:分类,命名和识别。分类以有序的方式群体群体,而命名法则涉及命名这些生物,要求国际协议以持续使用。命名法的更改可能会引起混乱,并受到国际商定的规则。在临床实践中,微生物学家主要专注于根据商定的分类系统识别分离株。这些组成部分以及分类法构成了与进化,遗传学和物种有关的系统学的总体学科。原生动物,真菌和蠕虫是根据卡尔·冯·林纳(Carl vonLinné)开创性工作后的标准规则分类和命名的。大类(阶级,秩序,家庭)进一步分为由拉丁二项式指定的单个物种。细菌表现出比所有其他细胞寿命的多样性更大,这使刚性分类具有挑战性。识别主要是通过基于密钥的系统来实现的,该系统基于生化性能测试系统的生长或活动来组织细菌性状。有些测试明确鉴定了属或物种,例如葡萄球菌属的过氧化氢酶产生。和细胞色素c由铜绿假单胞菌C。其他特征可能是单个物种独有的,将它们与具有相似生化谱的人区分开来。某些细菌在实验室中不生长(麻风细菌,treponemes),需要遗传学方法鉴定。如图它们可能构成一个属。随着遗传分析技术变得越来越容易获得,它们和其他快速分析方法正在取代传统的生化方法以识别。细菌分类中使用的分类等级包括王国(原核),分区(Gracilicutes),阶级(Betaproteobacteria),订单(Burkholderiales),家庭(Burkholderiaceae),属(Burkholderia)(Burkholderia)和物种(Burkholderia cepacacia)。通过DNA同源性分析将一些属(例如动杆菌)细分为基因组物种。细菌和病毒的分类构成了挑战,这是由于表型测试在区分某些基因组物种时的局限性。当前方法识别物种复合物,这些物种复合物使用多重分类学方法分为基因组群。例如,头囊菌络合物包括从植物病原体到人类病原体的各种生物。尽管没有普遍接受的分类系统,但Bergey的手册被广泛用作权威来源。国际系统细菌学委员会控制细菌命名法,并在《国际系统和进化微生物学杂志》中发布批准的细菌名称清单。病毒由国际病毒分类学委员会(ICTV)归类,并在病毒学档案中发表。在细菌分类中,主要组以基本特征(例如细胞形状,革兰氏染色反应和孢子形成)区分。属和物种通常通过发酵反应,营养需求和致病性等性质进行区分。不同字符的相对重要性通常是任意的,而Adansonian系统则使用考虑广泛字符的统计系数来确定菌株之间的关系程度。此方法可用于分类共享主要字符的较大分组中的菌株。通过评分多个表型特征,可以估计相似性或匹配系数,这些系数可以在计算机上计算以确定生物体之间相似性的程度。3.1,可以使用相似性矩阵或树状图来构建层次分类树。这种方法允许根据相似性水平(用虚线x和y表示)将生物体分离为属和物种。DNA中鸟嘌呤 - 胞嘧啶(G-C)碱基对之间的氢键强度大于腺嘌呤 - 胸腺胺(A-T)碱基对之间的强度,从而影响DNA熔化的温度。DNA序列以确定G+C含量,该含量在细菌属之间差异很大,但在物种中仍然相对一致。另一种分类方法涉及基于其DNA碱基序列的同源性进行分组。此方法利用了在受控冷却过程中的重新形态,并在互补区域之间产生混合配对。可以通过信使RNA(mRNA)结合研究获得有关相关性的遗传证据。尽管具有不同G+C比的生物不太可能显示出明显的DNA同源性,但具有相似或相同的G+C比的生物可能不一定具有同源性。系统发育相关性。已经开发了一种实时PCR方法来估计G+C含量。核糖体RNA(rRNA)的结构似乎在进化过程中是保守的,反映了系统发育关系。核苷酸测序相对简单,并导致了许多在线医学上重要的细菌物种的DNA序列的可用性。注意:我应用了“添加拼写错误(SE)”方法,其中有10%的概率引入错误。如果您要我以不同的方式重塑它,请让我知道!在此处给定文章的分枝杆菌物种鉴定对于理解其系统发育关系至关重要。尽管rDNA序列中的高相似性(> 97%),但可以使用Microseq(Applied Biosystems)等商业系统来区分不同的物种。但是,核糖体基因可能无法提供足够的变化来区分紧密相关的物种。替代候选基因(例如RECA)已被探索,并且似乎有望用于系统发育分析。在系统发育研究中也使用了其他家政基因,包括RPOB,GROEL和GYRB。这些基因定义了与RRNA基因观察到的基因一致的进化树。分类法的主要目标是促进在临床和公共卫生环境中的个人和团体的有效管理。然而,由于基因组序列数据揭示了微生物之间的相互关系,因此对与基本理解保持一致性是必要的。表3.1根据共享特征概述了简化的分类方案。门A(属)是正确的。这些群体已与最近确定的系统发育命名法对服。可以通过补充测试,有时在物种水平上进一步识别生物。形态标准足以鉴定原生动物,蠕虫和真菌。The classification of cellular micro-organisms is as follows: Eukaryotes: Protozoa - Sporozoa Plasmodium, Isospora, Toxoplasma, Cryptosporidium Flagellates Giardia, Trichomonas, Trypanosoma, Leishmania Amoebae Entamoeba, Naegleria, Acanthamoeba Other: Babesia, Balantidium Fungi: Mould-like Epidermophyton, Trichophyton, Microsporum, Aspergillus Yeast-like Candida Dimorphic Histoplasma, Blastomyces, Coccidioides True yeast: Cryptococcus Prokaryotes: Bacteria: Actinobacteria (High G+C Gram positives) - Actinomyces, Streptomyces, Corynebacterium, Nocardia,分枝杆菌,微球菌(低g-c gram阳性) - 李斯特菌,芽孢杆菌,梭状芽孢杆菌*,乳酸杆菌*,Eubacterium*革兰氏阳性杆菌,杆菌,芽孢杆菌,芽孢杆菌* Enterococcus Gram-negative cocci: Veillonella*, Mycoplasma Proteobacteria (a very large group with 5 sub-divisions) - Neisseria, Moraxella Gram-negative bacilli: Enterobacteria – Escherichia, Klebsiella, Proteus, Salmonella, Shigella, Yersinia Pseudomonads – Pseudomonas, Burkholderia, Stenotrophomonas Haemophilus, Bordetella, Brucella, Pasteurella Rickettsia, Coxiella Gram-negative curved and spiral bacilli: Vibrio, Spirillum, Campylobacter, Helicobacter Bacteroidetes - Bacteroides*, Prevotella* Borrelia, Treponema, Brachyspira, Leptospira衣原体衣原体这些单细胞生物是非斑型生物的,具有独特的核和细胞质。它们的大小从直径2-100 µm变化,其表面膜的复杂性和刚度有所不同。有些物种在内部捕获食物颗粒,而另一些物种则以细菌为食。原生动物被认为是最低的动物生命形式,它通过二元裂变或多重裂变无性繁殖。某些鞭毛原生动物与光合藻类密切相关。最重要的医学原生动物组包括Sporozoa,Amoebae和鞭毛。这些生物具有相对刚性的细胞壁,可能是腐生的或寄生的。霉菌随着分支丝的生长而生长,称为菌丝,形成了称为菌丝体的网状作品。通过形成从营养或空中菌丝体发展的性和无性孢子来繁殖。酵母是卵形细胞,通过萌芽并形成性孢子无性繁殖。二态真菌在人造培养中产生营养菌丝体,但在感染病变中类似酵母。主要的细菌组通过微观观察到其形态和染色反应来区分。革兰氏阴性程序将细菌分为两个伟大的分区:革兰氏阳性和革兰氏阴性细菌。然而,较旧的分类系统与较新的基于DNA序列的系统发育分类之间的关系是复杂的且仍在发展的。随着细菌组之间的系统发育关系开始解体,出现异常。文本描述了根据其形态学特征和染色反应对细菌和病毒进行分类的各种组。尽管如此,在临床实验室中采用的实际鉴定方案很大程度上取决于细菌的形状革兰氏阳性还是阴性,杆菌或球菌的形状,以及它们在有氧或厌氧上生长的能力。医学上有意义的细菌的主要系统发育组包括静脉细菌,其革兰氏阳性具有较高的G+C含量,具有丝状生长和菌丝体的产生; Firmicutes,一组低的G+C革兰氏阳性细菌,其中包括细菌,球菌和孢子形成器;蛋白质细菌,一大群革兰氏阴性细菌;细菌植物,革兰氏阴性厌食症;螺旋体,其特征是带有内部鞭毛的螺旋形细胞;衣原体,严格的细胞内寄生虫产生抗生素并具有非常重要的病原体。其他值得注意的组包括放线菌,链霉菌,分枝杆菌,诺卡氏菌,corynebacterium,链球菌,葡萄球菌,分枝杆菌,尿不质质,叶绿体,veillonella,veillonella,veillonella,gram阳性孢子形成的孢子形成杆菌和近亲,可能会变成gram- cortridium-new cortridiul cortridur cortriver cortridge cortridge cortridg corlam-infram-negam-inform-Gram-ne Gram-ne Gramne。例如,梭状芽胞杆菌的末端孢子具有独特的球形形状。革兰氏阳性的非孢子芽孢杆菌,包括甲ip骨和乳杆菌,倾向于在链或细丝中生长。相反,一些细菌具有使运动能力的鞭毛,例如李斯特菌。细菌可以根据其细胞壁组成,包括α-肾上腺细菌(包括人力赛组和布鲁氏菌),以及贝贝氏菌,包括静脉和伯克霍尔德里亚。尽管具有优势,但核酸测定并非没有局限性。此外,gamaproteobacteria包括大肠杆菌等肠杆菌,以及假单胞菌和军团菌。一些细菌的独特特性(例如弯曲的颤音,包括弧形霍乱)是值得注意的。divaproteobacteria群体在医学上并不显着,而Epsilonproteobacteria包括螺旋杆菌和弯曲杆菌,它们表现出螺旋形状。革兰氏阴性的非腐蚀性厌氧菌(如杆菌和prevotella)以其细长的柔性螺旋而区别。病毒,重点是它们对宿主细胞复制的依赖。某些病毒可能会包裹在脂蛋白中,而另一些病毒缺乏该外层。提出了一个分类系统,根据其遗传物质和衣壳结构对病毒进行分组。引起人类疾病的主要病毒类型包括RNA病毒,例如流感,paramyxoviruse和Flaviviviruses,以及picornaviruses和paciviruses。许多类型的病毒,包括艾滋病毒,HTLV和疱疹病毒会导致人类疾病。DNA病毒,例如痘病毒,轮状病毒和腺病毒,也感染了人。微生物学家在识别细菌时由于精确识别所需的耗时过程而面临挑战。通常,它们依赖于显微镜和培养物等简单方法,可以通过其他测试进行推定识别来支持。但是,这些方法通常至少需要24小时,因此在开始识别之前必须获得单个分离株的纯培养。与文化方法不同,非文化检测技术(例如抗原或基于核酸的检测)没有需要纯培养的缺点,但可能具有特异性的局限性。形态和染色反应可以作为将未知物种置于其适当的生物群中的初步标准。诸如革兰氏阴性,深色地面照明和阴性染色之类的技术可用于观察细菌形态,运动性和胶囊形成。在某些情况下,病理标本中某些生物体的微观特征可能足以进行假定的鉴定,例如痰液中的结节芽孢杆菌或渗出液中的T. pallidum T. pallidum。但是,许多细菌具有相似的形态特征,需要进一步测试以区分它们。固体培养基上殖民增长的出现还可以提供特征信息,包括菌落大小,形状,高程和透明度。微生物生长和特征的变化,包括透明度,不透明和颜色,可能会显着影响结果。生长所需的条件范围特定于某些生物,有些需要氧气,其他厌氧环境,而另一些则对二氧化碳水平或pH值敏感。为了区分相似的物种,可以采用评估代谢差异的测试,例如产生特定碳水化合物的酸性和气态终产物的能力。但是,现在许多实验室都使用了结合简单性和准确性的市售微磨合。此过程导致可见细菌生长的抑制作用。Some common tests used in identification include: - Production of indole or hydrogen sulphide - Presence of oxidase, catalase, urease, gelatinase, or lecithinase enzyme activities - Utilization of various carbon sources Traditionally, these tests have been performed individually according to standard guidelines.套件也可用于特定的生物组,例如肠杆菌和厌氧菌。在某些情况下,可以使用更先进的程序来分析代谢产物或全细胞脂肪酸。A fully automated system using high-resolution gas chromatography and pattern recognition software is widely used, allowing for the rapid identification of various bacterial species.Mass spectrometry also holds promise for rapid identification through matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry.由于细菌的多样性和复杂性,对细菌的检测和鉴定可能具有挑战性。Many organisms may not grow in culture, or they may require specialized nutrients, making traditional methods time-consuming and labor-intensive.然而,核酸技术的进步彻底改变了该领域,提供了更灵敏和快速的检测方法。Commercially available systems, including PCR, transcription-mediated amplification, and hybridization with specific probes, can identify a wide range of bacterial species with high accuracy.These technologies enable the detection of multiple species simultaneously, making them ideal for epidemiological investigations and antimicrobial susceptibility testing.此方法允许进行定量和形态评估。污染,操作员技能,底漆设计以及标本中抑制性化合物的存在都会影响结果。对这些结果的解释需要仔细考虑生物体的自然栖息地和共生主义的潜力。The development of new technologies, such as peptide nucleic acid (PNA) assays, holds promise for even more rapid and sensitive detection methods.These techniques use PNA molecules with DNA binding capacity to detect and identify bacterial species on microscope slides, and can be amplified using PCR to accelerate testing times.也已经开发出高密度寡核苷酸阵列,从而可以同时分析数千种不同的探针。This enables researchers to quickly identify specific genetic markers associated with antimicrobial resistance, paving the way for more targeted treatment strategies.Recent advancements include DNA sequencing, strain genotyping, and identifying gene functions, as well as locating resistance genes and changes in mRNA expression.一种创新的方法涉及在Eppendorf管中开发的选定基因靶标的阵列。The chip embedded in the tube contains optimized sets of oligonucleotide probes specific to certain organisms or antimicrobial resistance genes.这允许自定义单个细菌或组的芯片。从样品制备到检测的测定过程在单个管中在6-8小时内完成。实时PCR已广泛开发,使用荧光在单个反应管中结合了扩增和检测。该系统比常规PCR具有显着优势,包括速度,简单性和减少手动程序。基于荧光的方法可以检测DNA产物或通过与荧光标记的探针杂交提高特异性。对靶DNA的定量也是可能的,可以估计样品中的病毒或细菌数。 此外,针对16S核糖体RNA的荧光原位杂交(FISH)已用于直接在临床标本中检测细菌,而无需培养。 可以通过血清学反应来鉴定微生物的种类和类型,这些反应依赖于特有的特定物种或类型的抗体或类型的抗体,这些抗体以特征性的方式与微生物反应。 抗体在检测细菌产生的毒素和抗原以及鉴定特定病毒方面起着至关重要的作用。 基于乳胶的试剂盒广泛用于血清学组和毒素检测。 在ELISA中,特异性抗体附着在塑料孔上,并添加了测试抗原。 通过添加更特异性的抗体检测到抗原的存在,并用启动颜色反应的酶标记。 ELISA方法可以反向使用以定量检测抗体。 在Mac-Elisa中,纯化的抗原被吸附到井中,并添加了测试血清。 任何IgM与捕获试剂结合,并添加纯化的抗原以用标记的抗体检测。 某些病毒,例如流感,在红细胞上充当桥梁的受体,形成可见的团块。 但是,这种方法缺乏可重复性。对靶DNA的定量也是可能的,可以估计样品中的病毒或细菌数。此外,针对16S核糖体RNA的荧光原位杂交(FISH)已用于直接在临床标本中检测细菌,而无需培养。可以通过血清学反应来鉴定微生物的种类和类型,这些反应依赖于特有的特定物种或类型的抗体或类型的抗体,这些抗体以特征性的方式与微生物反应。抗体在检测细菌产生的毒素和抗原以及鉴定特定病毒方面起着至关重要的作用。基于乳胶的试剂盒广泛用于血清学组和毒素检测。在ELISA中,特异性抗体附着在塑料孔上,并添加了测试抗原。通过添加更特异性的抗体检测到抗原的存在,并用启动颜色反应的酶标记。ELISA方法可以反向使用以定量检测抗体。在Mac-Elisa中,纯化的抗原被吸附到井中,并添加了测试血清。任何IgM与捕获试剂结合,并添加纯化的抗原以用标记的抗体检测。某些病毒,例如流感,在红细胞上充当桥梁的受体,形成可见的团块。但是,这种方法缺乏可重复性。Haemagglutinins can be detected in tissue culture, and red cells can be coated with specific antibodies to agglutinate in the presence of homologous virus particles.荧光染料可用于染色组织或生物体,从而在紫外线下可视化。Antibody molecules can be labeled with fluorochrome dyes, enabling direct immunofluorescence procedures for highly sensitive antigen identification.该技术将抗体技术与PCR方法相结合,以增强抗原检测能力。分子生物学中的一种新方法涉及将DNA分子与抗原抗体复合物联系起来,从而产生特定的结合物。此附件允许通过PCR扩增,验证抗原的存在。免疫-PCR的增强灵敏度超过ELISA的105倍,因此检测到只有580个抗原分子。细菌种群表现出不同的结构,从高度多样化到非常相似。Recombination frequency is the primary determinant of population structure, with some species experiencing high recombination rates and others exhibiting rare recombination events.Species such as Neisseria gonorrhoeae are naturally transformable, displaying high recombination frequencies, while Salmonella enterica populations exhibit low recombination rates.细菌克隆可能显示出瞬态或持久特征。Panmictic与克隆人群的概念突出了这两种类型之间的繁殖,重组,等位基因排列和选择性压力的差异。In each family lie many genera of each type.键入分离株可以与参考标记,识别细菌物种中的菌株和分离株进行比较。区分类似菌株的能力在追踪社区或医院环境中感染的来源或传播方面具有重要意义。已经开发了各种键入方法来帮助这一过程,这可能涉及从相同起源菌株之间识别较小的差异。尽管单个打字方法可以证明相同的响应,但这不是两种菌株相同的结论性证据。但是,使用多种打字方法大大提高了相似性的置信度。键入技术可以在不同的流行病学水平上应用,包括微流行病学,宏观流行病学和种群结构分析。从键入中得出的数据可以通过识别共同或点源,区分混合应变感染以及识别再感染与复发与复发来帮助控制感染。一些方法还有助于识别与疾病相关的特定类型,例如大肠杆菌O157和溶血性尿毒症综合征。为了使方法被认为是可靠的,必须在实验室环境和临床上可以重现。在流行病学研究的背景下,首选多种键入方法,因为它们可以针对不同的特征。这些包括生物化学测试,这些测试定义了物种内的生物型,抗性分型检测对化学物质敏感性的变化以及基于营养需求的生长需求的辅助分型。可以使用此方法分析质粒和染色体DNA。此外,许多细菌的表面结构都是抗原性的,可以使用针对它们提出的抗体将分离株分为定义的血清型。物种可以根据其独特特征分为几种抗原类型。对于某些物种,血清分型是一种识别和区分不同菌株的高效方法。在其他情况下,抗原表位的保存使血清型对流行病学目的的有用程度降低。例如,沙门氏菌的物种可以通过其体细胞和鞭毛血清型来定义。研究表明,囊抗原可能在某些生物的致病性中起作用,许多疫苗通过刺激对这些抗原的抗体来起作用。噬菌体键入是一种用于识别和区分细菌菌株的方法。这涉及使用特定噬菌体的凝集或降水反应,如果适当地适应,这可能具有很高的歧视性。但是,某些噬菌体集缺乏稳定性会导致广泛的噬菌体组,而不是定义的类型。此外,控制噬菌体分型结果解释的关键因素是歧视和可重复性。噬菌体与细菌之间的相互作用是一个复杂的过程,涉及吸附,DNA注射以及裂解或复制。裂解或有毒的噬菌体可以在复制循环结束时裂解宿主细胞,从而释放可能感染相邻细胞的新噬菌体颗粒。但是,其有效性取决于噬菌体的适应和系统的稳定性。噬菌体键入已用于包括微生物学和流行病学在内的各个领域,以识别和跟踪细菌菌株。尽管存在这些局限性,但噬菌体打字仍然是理解不同细菌菌株及其特性之间关系的重要工具。只有在两个强烈的裂解反应表现出两种不同的菌株时,才能识别出两种不同的菌株。细菌素是大多数细菌物种产生的自然存在的抗菌物质,主要靶向与生产菌株同一属内的菌株。通过分析产生的细菌素的光谱或对标准面板细菌素的敏感性,细菌素键入可以定义不同类型的细菌。蛋白质组学分析,涉及具有强洗涤剂的丙烯酰胺凝胶中的凝胶电泳,也可以通过可视化数千种蛋白质并比较分离物之间的带模式来鉴定细菌物种。另外,研究人员已使用凝胶电泳来分析代谢酶,可以使用特定底物检测到该酶,用于物种内的克隆分析。限制性核酸内切酶是在特定序列识别位点切下DNA的酶。这些切割的频率取决于寡核苷酸序列,限制位点的频率以及所检查的物种的G+C含量的百分比。频繁切割的核酸内切酶产生许多小片段,可以通过琼脂糖凝胶中的常规电泳解决,并通过用染料染色检测。通过引入脉冲或在电场方向上变化,可以分开碎片至10 MB。相比之下,不经常的切割酶产生的大型DNA片段需要脉冲场凝胶电泳(PFGE)进行分离。该技术涉及将细菌包裹在琼脂糖塞中,用蛋白酶K酶消化细胞,然后用酶消化DNA。CORTOUR夹具均匀的电场(Chef)设备通常用于PFGE,并具有在六角形阵列中排列的24个电极。运行时间通常在30到40小时范围内,尽管已经描述了较短的协议。几个因素影响了这些分析的结果,包括正在检查的DNA类型,酶和反应条件的选择以及所使用的设备质量。DNA样品的质量和浓度,琼脂糖凝胶电压和脉冲时间,缓冲液强度和温度会影响脉冲场凝胶电泳(PFGE)的结果。虽然解释PFGE曲线可能是由于不同物种之间的带状模式的变化而具有挑战性的,但已通过Tenover确定了特定的标准以确定差异的重要性。通常,与显示剖面无差异的单个事件中的分离物被认为是无法区分的。一到三个频段差异的人密切相关。四到六个乐队可能表明可能的关系;七个或更多的差异表明不同的菌株。但是,该规则应谨慎应用,因为即使在同一克隆的成员之间,某些物种也会表现出显着差异。Pearson系数是另一种常用的方法,具有不需要定义特定带位置的优势。可以使用计算机辅助分析软件包来计算菌株之间相似性的系数,例如jaccard和骰子系数,这些系数使用配置文件中的一致频段来确定百分比相似性。经常使用85%相似性的截止点,但应通过实验相关且无关的应变集设置。DNA探针可以根据克隆的特异性,随机序列或通用序列检测靶DNA中的限制位点异质性。rubotyping检测rDNA基因基因座的变化,并已普遍应用于各种物种。其他常用的探针是可能定义种群克隆结构的插入序列。PCR(聚合酶链反应)是一种允许在受控条件下放大特定DNA序列的技术。可以通过使用PCR的重复放大循环来制作由特定寡核苷酸引物定义的基因组区域的多个副本。该方法已广泛用于DNA指纹和键入,利用DNA分子中的可变区域,例如串联重复区域的可变数量或具有限制性核酸内切酶识别序列的区域。两种方法都有局限性,这是由于错误启动,不同的带强度以及电泳迁移差异引起的可重复性问题。基于重复序列的PCR(REP-PCR)索引在整个基因组中多个重复序列中的变化,而自动化的REP-PCR系统对应变键入显示了有望,并且可以提供与PFGE相似的歧视。狼在can属中,而狐狸则处于喧嚣中。放大的片段长度多态性结合了限制性核酸内切酶消化与PCR,以优化基因组之间单碱基对差异的可重复性和分辨率。该技术使用核苷酸测序来分析管家基因,该基因慢慢多样化,不受选择性的作用。多焦点序列分型(MLST)可以视为确定的基因分型。但是,MLST可能对诸如结核分枝杆菌等高度均匀的物种没有效。为了增加歧视,由于环境变化,毒力相关的基因提供了较高的序列变化,因此已经针对了毒力相关的基因。通过PCR扩增基因间区域,并测序了500 bp的内部片段以识别等位基因多态性。多焦点限制输入引入了放大管家基因的限制消化,从而消除了对测序的需求。可变数字串联重复序(VNTR)是拷贝数变化的短核苷酸序列,可用于快速且可再现的键入。识别其他遗传基因座可以提供进一步的见解,但随着时间的流逝,它们的稳定性仍然存在争议。DNA测序技术的最新进展使得分析整个基因组序列成为可能,从而可以更精确的比较和细菌的键入。这种方法涉及生成可以组装并与先前分离株进行比较的短核苷酸序列读取。与这些高级分析相关的成本与传统方法变得越来越具竞争力。这样的分析可以在同期和历史分离株之间建立进化关系,从而对细菌进化有更明确的理解。此外,这项技术通过提供明确的流行病学信息并确定有助于抗生素耐药性和抗原选择压力来转化医学细菌学的重要潜力。资料来源:Barrow Gi,Feltham RKA,编辑;加里斯总经理,编辑; Kaufmann我; Murray PR,Baron EJ,Jorgensen JH,编辑;欧文·RJ; Schleifer KH; Spratt BG,Feil EJ,Smith NH; Tenover FC,Arbeit Rd,Goering RV; Van Regenmortel MHV,Fauquet CM,Bishop DHL,编辑; Woese Cr。分类类别是称为分类单元的层次组,其中包含一小部分物种,该物种来自一个相对较新的共同祖先。可以在下面可视化整体层次结构以供参考:尽管研究不同生物体的科学家在分类方案中有所不同,但属背后的一般概念是它代表物种祖先相关的物种,并且与其他属不同,不包括不必要的物种。确定这在于每个研究者,但是这些一般指南在属属方面保持分类相当狭窄。属属的分类单元通常包括群体之间可识别的身体形式。例如,Felidae和Canidae分别代表类似猫的生物和类似狗的生物。最后一步,物种定义了在连续单位中共同繁殖的人群和群体。在一起,这些名字告诉您有关生物体的很多信息。在大多数情况下,由于遗传,行为或形态学差异,不同的属将不会繁殖。Carl Linnaeus通过他的生物生物命名计划(二项式命名法)普及了“属”一词,尽管他对属的定义与我们的现代观点有所不同,但在二项式命名法中使用通用epithets在二项式术语中的使用仍在继续。通用称呼是二项式命名法中描述有机体所属属的动物名称的两个单词。第二个单词或特定的称呼描述了有机体所属的生物或物种更紧密相关的群体。通过了解一个人也知道家庭,秩序和所有其他分类分类。由于分层群体是由生物之间的相似性安排的,所以这些关系告诉了我们很多有关单个动物的信息。知道该物种可以告知我们动物与该属中其他动物的独特性。例如,Honey Badger具有科学名称Mellivora Capensis。有时,属可能包含数百种物种,尤其是在鱼类和无脊椎动物中。这种品种具有误导性,因为它应该反映进化。进化多样性决定了属内生物的数量。如果许多物种随着属的传播而出现,将会有许多物种。相反,如果只有一个物种幸存,则只有一个物种。分类分类是一个持续的过程,每天都描述了新的属。一些新发现的生物从未被命名,而另一些有机体则根据DNA分析重新分类。通过分析DNA,比较性状并提出系统发育,科学家假设最可能的进化进展。这将为命名惯例提供信息,并确定哪些物种可以成为独特的属。物种代表属内生殖分离并与其他群体独特的群体。家庭是分层分类中属的分类单元。分类单元是指具有相似特征的群体。两条鱼一起游泳可能不会繁殖,而是具有类似的特征,与其他任何海洋鱼不同。如果它们可以杂交,则将被视为物种。北极熊和棕熊在同一属中是不同的物种,但仍可以成功繁殖。这是因为它们占据了独特的生态位,很少彼此遇到繁殖。生态障碍可以阻止它们自然繁殖,即使它们的后代是可行的。随着气候变化耗尽冰盖,可以将北极熊推向较低的纬度,并可能与棕熊杂交。科学家辩论是否应基于进化连接和物理特征将新物种添加到属中。如果两组共有共同的血统,则它们应属于同一属,即使它们在细胞外基质产生等特征上有所不同。在Fakus细菌的情况下是一种具有相似DNA但缺乏定义该属的独特基质的新物种,分类学家必须权衡多个领域的证据。通过分析解剖学,行为和遗传数据,科学家可以重建生物体之间的关系,并就分类做出明智的决定。