XiaoMi-AI文件搜索系统
World File Search System试验产品

第一阶段研究的最新结果
基线值是筛选期间不同日期的 2 个样本和研究第 1 天给药前的 1 个样本的平均值。仅显示队列中所有患者完成的访问。虚线表示目标最小减少量。星号表示自上次 2023 年 2 月 17 日数据截取以来开始额外的持续随访。SD,标准差。本演示文稿包含尚未获得监管机构批准的试验产品的数据。
食品和营养研究所-Dost
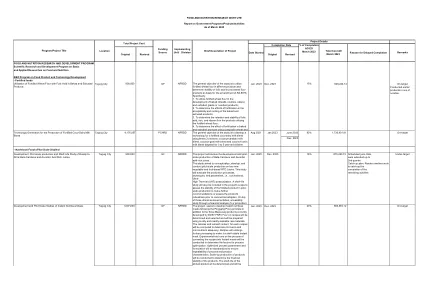
taguig City该项目旨在根据Pinggang Pinoy原理开发即时的营养餐,此外还有DOST FNRI目前开发的三种餐用品产品。将确定和选择四(4)个食谱,并将使用本地且随时可用的原材料制备。将计算每个食谱的卡路里和营养含量,以确定其宏观和微量营养素的充分性。食谱将进行进一步的处理,以使其成为货架稳定的即时餐。实验试验将在将食谱转换为即时餐的过程中进行,以确定过程优化的因素。优化的过程参数和公式将被标准化,以确保过程和产品特征的重复性。将进行产品的扩大生产,以确定产品的财务稳定性。将确定试验产品的保质期,将是

印度的临床试验和生物医学研究
临床试验是一种研究,旨在开发新的测试和治疗方法,以评估其对人类健康的影响。 3 对医疗干预的结果,即对人类志愿者的研究产品或药物(“新药”)进行评估。 试验产品可以是药物、疫苗、医疗器械、外科和放射学程序、行为治疗、预防保健、细胞和生物制品,这些产品是在该国获得上市许可后制造的。 4 临床试验是根据利益相关者(患者、医生和实验主持人)的需求为每种新药进行独特而周到的策划。 5 试验程序经过审查,只有在获得批准后才能开始试验。 6 因此,临床试验是对人类受试者(无论是患者还是非患者志愿者)的药品进行系统研究,以发现或验证临床、药理学(包括药效学/药代动力学)和不良反应,目的是在产品在印度上市之前确定其安全性和有效性。 7

观点文章赤道几内亚疟疾疫苗倡议:从启动临床研究平台到制定疟疾消除计划
PfSPZ 疫苗(Sanaria Inc.,马里兰州罗克维尔)。反过来,疫苗开发计划正在建设人力资本和物质能力。EGMVI 建立了监管和道德监督,以确保遵守国际协调会议和良好临床实践,这是赤道几内亚历史上首次进口试验产品、获得道德批准和开展临床试验。EGMVI 已在赤道几内亚完成了三次疫苗试验、在坦桑尼亚完成了两次疫苗试验和一项疟疾发病率研究,并启动了一项 2,100 名志愿者临床试验的准备工作。人员正在国外接受高级学位培训,并接受了良好临床实践和特定方案方法的培训。新设施为国家研究机构奠定了基础。在这个富有远见、雄心勃勃的公私伙伴关系中,生物医学研究和开发正在促进赤道几内亚的重大改进。 EGMVI 计划使用 PfSPZ 疫苗与标准疟疾控制干预措施来消灭比奥科岛的 Pf 疟疾,并成为其他地方消灭疟疾运动的潜在典范。

工业指南
I. 引言 人类基因治疗旨在修改或操纵基因的表达或改变活细胞的生物学特性以用于治疗。我们,FDA,为您,人类基因治疗新药临床试验申请 (IND) 的申办者,提供有关在 IND 中提交的化学、制造和控制 (CMC) 信息的建议。本指南旨在告知申办者如何提供足够的 CMC 信息,以确保试验产品的安全性、特性、质量、纯度和强度(包括效力)(21 联邦法规 (CFR) 312.23(a)(7)(i))。本指南适用于人类基因治疗产品和包含人类基因治疗与药物或器械的组合产品 1。 2 本指南最终确定了 2018 年 7 月发布的同名指南草案,并取代了 2008 年 4 月发布的文件“FDA 审评人员和申办方指南:人类基因治疗临床试验新药申请 (IND) 的化学、制造和控制 (CMC) 信息的内容和审评”(2008 年 4 月指南)。自我们发布 2008 年 4 月指南以来,基因治疗领域取得了迅速发展。因此,我们正在更新该指南,为您提供有关基因治疗 IND 的 CMC 内容的最新 FDA 建议。本指南的组织结构遵循 FDA 关于通用技术文件 (CTD) 的指南结构。有关 CTD 的信息可在 FDA 的行业指南中找到:“M4Q:CTD - 质量”(参考文献 1)。有关提交电子 CTD(eCTD)的信息,请参阅 FDA 网站 https://www.fda.gov/drugs/electronic-regulatory-submission-and-review/electronic-common-technical-document-ectd 。

实现基因组医学的未来
本演示文稿及其随附的口头评论包含有关我们当前期望的前瞻性陈述。这些前瞻性陈述包括但不限于与下列事项有关的陈述:我们候选产品和工程衣壳的治疗和商业潜力,包括 STACTM-BBB 释放治疗各种神经系统疾病的巨大潜力的能力,我们计划专注于表观遗传调控和衣壳工程,开发、获得监管部门批准并商业化治疗某些疾病的持久、安全、有效疗法的潜力以及此类疗法的时机、可用性和成本,使用 ZF、MINT 平台、SIFTER 和其他技术开发持久、安全、有效的疗法和衣壳的潜力,我们从合作中受益并获得开发和商业里程碑及特许权使用费的潜力以及任何此类收益和付款的时机,基因泰克完成临床开发、监管互动、制造和任何由此产生的产品的全球商业化的潜力,辉瑞对 giroctocogene fitelparvovec 计划的持续推进,包括辉瑞完成临床开发、监管互动、制造和任何由此产生的产品的全球商业化的潜力,预期从现有和新合作中获得的收入及其时间安排、为某些项目寻求合作伙伴或合作者的计划和预期、isaralgagene civaparvovec 符合 FDA 加速审批计划的可能性,包括在第 1/2 阶段 STAAR 研究中生成的数据是否足以支持任何此类批准;对于是否有更多数据支持 isaralgagene civaparvovec 的潜在 BLA 提交以及提交时间的预期;加快预期审批时间表并比之前预期更快地将 isaralgagene civaparvovec 带给患者的潜力; isaralgagene civaparvovec 预计的注册进展,包括我们寻求潜在合作伙伴的计划、有关我们财务资源的计划(包括其充足性)以及降低运营费用的计划、我们精简结构和未来潜在成本削减的影响、我们和我们的合作者进行正在进行的和潜在的未来临床试验的预期计划和时间表以及展示我们和我们合作伙伴的临床试验数据并提交监管文件、我们候选产品预计的后期开发进展(包括潜在的未来注册试验)、我们公司战略的执行、我们的产品线、其他目标的确定以及临床前项目向临床的进展、关键里程碑和催化剂,以及其他非历史事实的陈述。这些陈述并非对未来表现的保证,并且受难以预测的某些风险和不确定性的影响。我们的实际结果可能与所表达的结果存在重大不利差异。可能导致实际结果不同的因素包括但不限于不确定和昂贵的研发过程,包括临床前结果可能无法预示任何未来临床试验的风险、与宏观经济因素相关的风险和不确定性,包括由于持续的海外冲突、由于银行倒闭导致银行存款和贷款承诺的中断、对全球商业环境、医疗保健系统以及我们和我们的合作者的业务和运营的影响,包括临床试验的启动和运营;研发过程;临床试验的不确定时间和不可预测的结果,包括初步或初始临床试验数据是否代表最终临床试验数据,以及最终临床试验数据是否验证候选产品的安全性、有效性和耐用性;临床试验延迟、暂停和搁置对临床试验时间表和候选产品商业化的影响;多个监管机构对产品候选物的不可预测的监管审批程序;产品、产品候选物和衣壳的制造;已获批准产品的商业化;技术发展可能会取代我们和我们的合作者所使用的技术;我们或我们的合作者违反或终止合作协议的可能性;我们可能无法实现预期的合作效益;我们未来资本需求、财务表现和业绩的不确定性,我们缺乏资本资源来充分开发、获得监管部门批准和商业化我们的产品候选物,包括我们确保某些项目合作的能力、我们确保及时或完全推进临床前项目所需资金的能力;以及我们需要大量额外资金来执行我们的运营计划和持续经营,包括我们无法获得推进临床前和临床项目所需资金的风险,也无法持续经营,在这种情况下,我们可能被要求完全停止运营、清算全部或部分资产和/或根据适用的破产法寻求保护。我们和我们的合作伙伴无法保证能够开发出具有商业可行性的产品。这些风险和不确定性在我们截至 2023 年 12 月 31 日的财年 10-K 表年度报告中有更详细的描述,经 Sangamo 提交给美国证券交易委员会(“SEC”)的截至 2024 年 9 月 30 日的 10-Q 表季度报告和提交给 SEC 的未来报告补充。本演示文稿中包含的前瞻性陈述仅代表本演示文稿之日的观点,除适用法律要求外,我们不承担更新此类信息的义务。本演示文稿涉及正在进行临床前和/或临床研究且尚未获得任何监管机构批准上市的试验产品候选物。它们目前仅限于研究用途,并且对于它们在研究目的下的安全性或有效性不作任何陈述。任何关于安全性或有效性的讨论仅针对此处介绍的具体结果,可能并不代表监管机构最终认定的安全性或有效性。