机构名称:
¥ 1.0
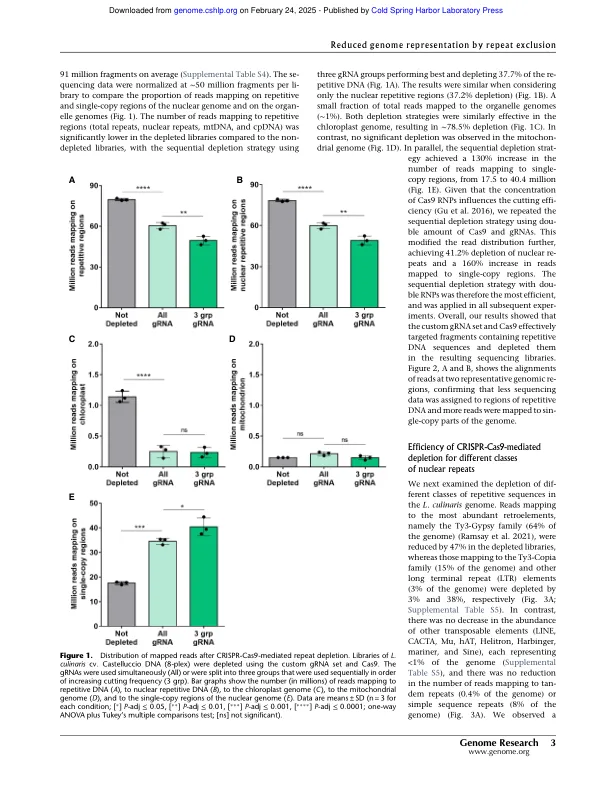
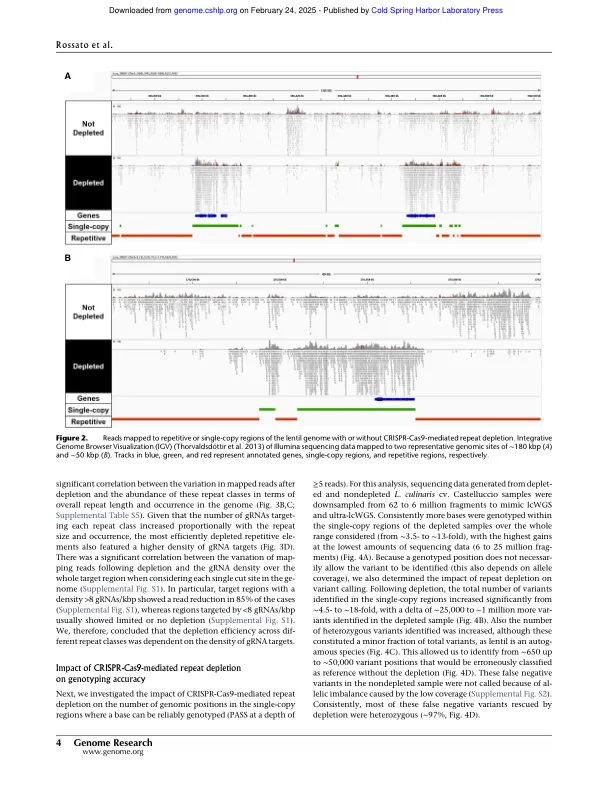
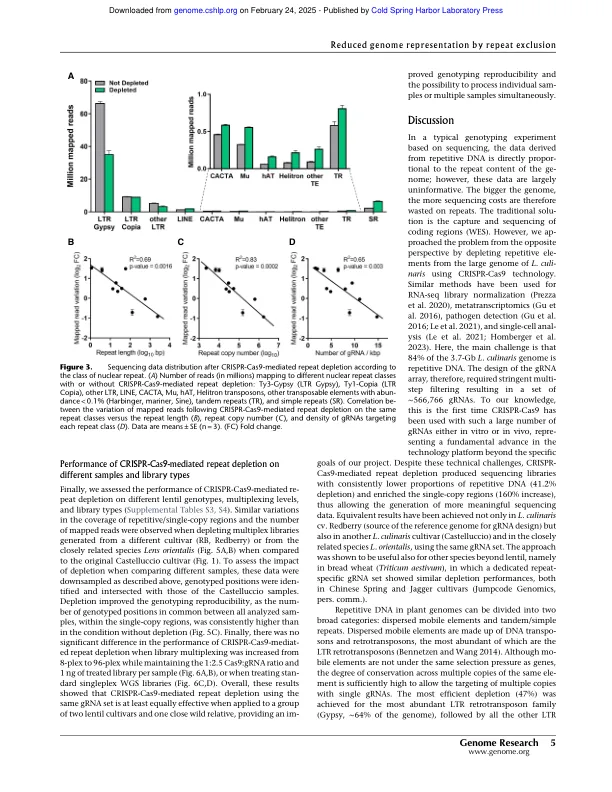
高通量基因分型能够对种群基因组学和全基因组关联研究中的遗传多样性进行大规模分析,这些研究结合了大量加入的基因型和表型表征。基于测序的基因分型方法由于较低的确定性偏差而逐渐替换传统的基因分型方法。然而,基于测序的全基因分型在具有较大基因组和高比例的重复性DNA的物种中变得昂贵。在这里,我们描述了CRISPR-CAS9技术在3.76-Gb基因组(镜头Culinaris)中耗尽重复元素,84%由重复序列组成,从而将测序数据集中在编码和调节区域(单子拷贝区域)上。我们设计了一组566,766个GRNA,旨在重复2.9英镑,排除了基于ATACSQ数据的重复区域重复的注释基因和推定的调节元素。新颖的耗竭方法去除了〜40%的读取映射到重复序列,从而将这些映射到单拷贝区域增加了约2.6倍。在分析2500万个片段时,与非部位的文库相比,测序数据中的重复对单个拷贝偏移增加了约10倍。在相同的条件下,我们还能够鉴定单拷贝区域中的遗传变异量增加了12倍,并通过挽救杂合变体的特征来提高基因分型精度,否则由于覆盖范围较低,否则会遗漏这些变体。该方法的执行方式类似,无论多路复用水平,文库类型或基因型,包括不同的品种和密切相关的物种(L. Orientalis)。我们的结果表明,CRISPR-CAS9驱动的重复耗竭将测序数据集中在单拷贝区域上,从而改善了大型和重复的基因组中的高密度和全基因组基因分型。
全基因组长阅读测序降采样及其对变化精度和回忆
主要关键词


相关文件推荐