XiaoMi-AI文件搜索系统
World File Search System断点

复杂的染色体重排中的断点对应于成熟精子的DNA的转座酶可访问区域
电气惊人用于捕获鳄鱼以执行常规管理程序。从福利点开始,电气令人惊叹必须引起动物的无意识。然而,没有信息有关电气令人惊叹是否引起尼罗河鳄鱼(Crocodylus niloticus)的无意识。该研究的目的是使用5通道参考脑电图分析来评估鳄鱼中电气惊人之前和之后的大脑活动,以确定意识。的行为指标和15个圈养鳄鱼的脑电图记录被捕获并使用功率谱密度分析在令人惊叹前后的功率频谱密度分析进行分析,然后以60 s的间隔,直到播放后5分钟。在湿颈上施加了5–7 s的标准化刻度170伏。无意识的定义是α波功率的降低和增长三角波功率的增加。无法评估三个脑电图。在12个鳄鱼中的6个中发现了无意识,平均持续120 s。脑电图波形振幅和滋补性癫痫发作的波形活性和行为指标的增加并不是可靠的无意识指标。进一步的研究应集中于提高电气惊人的效率和可靠性。

CRISPR-Cas9 介导诱导人类支气管上皮细胞发生大型染色体倒位
1. 通过 UCSC 基因组浏览器 ( https://genome.ucsc.edu/ ) 可获得用于设计两个 gRNA 的目标 DNA 序列。a. 选择感兴趣的基因组版本。在我们的例子中,使用的是“人类 GRCh38/hg38”。b. 根据已知的倒位断点 1 的位置,标记断点前 100-150 bp 到断点后 100–150 bp 范围内的基因组区域。例如,如果断点 1 位于 chr3:2,920,305,则在 UCSC 基因组浏览器搜索框中输入“chr3:2,920,205–2,920,405”以标记所需的染色体区域,然后单击“Go”。c. 在 UCSC 基因组浏览器工具栏上选择“查看”,然后单击“DNA”选项。d.在新窗口中,单击“获取 DNA”以获得准确的 DNA 序列。这是使用 CRISPOR 算法设计 gRNA 引物所需的序列(见下面的步骤 2a)。e. 对倒位的断点 2 重复步骤 1a-1d。2. 要设计 gRNA,请使用 CRISPOR 算法(http://crispor.tefor.net/):a. 输入从步骤 1d 获得的断点 1 的 DNA 序列。确保参考基因组与 UCSC 浏览器(步骤 1a)中使用的基因组相匹配,然后选择可通过转染载体编码的 Cas9 酶类型识别的 Protospacer Adjacent Motif (PAM)。如果转染载体表达 SpCas9,则选择 20 bp-NGG PAM 格式。单击“提交”以获得针对模板 DNA 的候选 gRNA 序列。b. CRISPOR 算法默认按特异性从高到低对候选 gRNA 序列进行排序,因为这是关键参数。从新页面上出现的候选 gRNA 列表中,选择具有最高麻省理工学院 (MIT) 和切割频率确定 (CFD) 特异性得分的指导序列(Doench 等人,2016 年;Hsu 等人,2013 年;Tycko 等人,2019 年)。这些分数根据以下方面评估候选 gRNA

使用 FUGAREC 检测长读转录组测序数据中的融合基因
摘要:融合基因是癌症治疗的重要靶点和生物标志物,临床需要准确检测融合基因的方法。RNA-seq被广泛用于检测活性融合基因。长读RNA-seq可以对mRNA全长进行测序,有望检测出短读RNA-seq无法检测到的融合基因。然而,长读RNA-seq的碱基调用错误率较高,在与基因组不一致的长读的断点附近可能会出现间隙序列。当出现间隙序列时,现有方法无法识别正确的融合基因或断点。为了解决融合基因检测中的这些挑战,我们引入了一种新算法FUGAREC(带间隙重新对齐和断点聚类的融合检测)。FUGAREC独特地将间隙序列重新对齐与断点聚类结合在一起。这种方法不仅增强了对以前无法检测到的融合基因的检测,而且显著降低了假阳性。我们证明 FUGAREC 在乳腺癌细胞系的模拟数据和测序数据上都具有很高的融合基因检测性能。

更改CALGARY区和南部区域的BCR :: ABL1定量测试
• As of May 2, 2024 all BCR::ABL1 RT-PCR quantitative testing (major and minor) will be transitioned to the Health Canada-approved Asuragen QuantideX qPCR BCR-ABL IS kit and Minor kit respectively, due to recent lack of availability and quality issues with the previously utilized BCR::ABL1 reagents (QIAGEN Ipsogen kits).新测定法检测到与先前测定的相同的成绩单变体,并根据通过IRIS建立的国际标准(用于主要断点)报告。新的主要断点测定法具有略有改善的转录检测下限(log 4.7降低 /%为0.002%;先验套件,log 4.5降低)。新的次要断点测定法具有提高的转录检测的下限(log 4.61降低 /%为0.0025%;先验套件,log 3.6降低)。

指南 - 抗菌治疗药物监测
1:使用单独的患者麦克风(如果有),或使用Eucast中可用的断点MIC,请参阅eucast:临床断裂点和抗生素剂量。如果病原体报告了对抗菌剂的中间敏感性 - 寻求专家建议(目标槽浓度可能更高)。由于对假单胞菌属的常规报告。对于某些药物,已包括这些靶标。2:使用高于20 mg/L的浓度,因为它们比断点麦克风高10倍以上,尽管毒性通常在50 mg/l以下不高。3:基于头孢洛齐浓度的目标浓度。

双内含子靶向 CRISPR-Cas9 介导的 AML RUNX1-RUNX1T1 融合基因破坏可在体内和体外有效抑制增殖并减小肿瘤体积
致癌融合驱动因子在血液癌症中很常见,因此是未来基于 CRISPR-Cas9 的治疗策略的相关靶点。然而,患者断点位置的变化对传统的断点靶向 CRISPR-Cas9 介导的破坏策略构成了挑战。在这里,我们提出了一种新的双内含子靶向 CRISPR-Cas9 治疗策略,用于靶向 5-10% 的新生急性髓系白血病 (AML) 中发现的 t(8;21),该策略可有效破坏融合基因,而无需事先确定断点位置。与非 t(8;21) AML 对照相比,在 RUNX1-RUNX1T1 双内含子靶向破坏后,AML t(8;21) Kasumi-1 细胞的体外生长率和增殖率分别降低了 69% 和 94%。此外,与对照组相比,注射了 RUNX1-RUNX1T1 破坏的 Kasumi-1 细胞的小鼠体内肿瘤生长减少了 69% 和 91%。这些发现证明了 RUNX1-RUNX1T1 破坏的可行性,在从被诊断为 AML t(8;21) 的患者身上分离的原代细胞中得到了证实。总之,我们证明了 AML t(8;21) 中双内含子靶向 CRISPR-Cas9 治疗策略的原理验证,而无需精确了解断点位置。
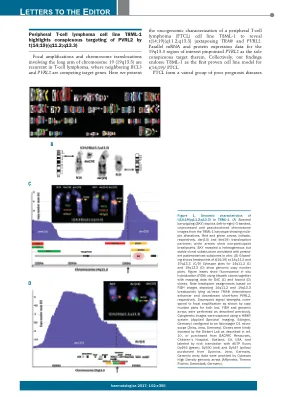
周围T细胞淋巴瘤细胞系T8ML-1突出显示了T(14; 19)(Q11.2; Q13.3)
图1。T(14; 19)(Q11.2; Q13.3)在T8ML-1中的基因组特征。 (a)光谱核分型(天空)描绘了来自T8ML-1核型的G带,未加工和伪色彩的染色体图像,显示了多个PLE改变。 红色和绿色箭头分别表示DER(14)和DER(19)易位伙伴;白色箭头显示非参与者断点。 天空揭示了与持续存在的患者衍生的亚克隆一致的异质但稳定的克隆下结构。 (b)G频段显示了14Q11.2和19Q13.3的T(14; 19)的断点。 (c/d)14q11.2(c)和19q13.3(d)的Cytoscan图显示了基因组拷贝数图。 图插图显示了使用Tilepath克隆以及映射BAC(C)和Fosmid(D)克隆的映射数据的荧光原位杂交(FISH)。 请注意基于鱼图像的断点分配,描绘了14q11.2和19q13.3分别位于Tra@ dowr@下游增强子和下游短形式PVRL2的断点。 差异信号强度符合焦点扩增,如两个基因座的拷贝数图所示。 如前所述,进行了鱼类和基因组阵列。 使用HISKY系统(Applied Spectral Imaging,Edingen,Germany)捕获了细胞遗传学图像,该系统配置为AxioImager D1 Micro-Scope(Zeiss,Jena,Germany)。 如参考文献中所述,Siebert Lab友好地捐赠了克隆。 10,或从美国加利福尼亚州奥克兰市的BACPAC资源,儿童医院购买,并由Nick Translation用Dutp Fluors Dy495(绿色),DY590(RED)和DY547(黄色)(黄色)(黄色)购买。T(14; 19)(Q11.2; Q13.3)在T8ML-1中的基因组特征。(a)光谱核分型(天空)描绘了来自T8ML-1核型的G带,未加工和伪色彩的染色体图像,显示了多个PLE改变。红色和绿色箭头分别表示DER(14)和DER(19)易位伙伴;白色箭头显示非参与者断点。天空揭示了与持续存在的患者衍生的亚克隆一致的异质但稳定的克隆下结构。(b)G频段显示了14Q11.2和19Q13.3的T(14; 19)的断点。(c/d)14q11.2(c)和19q13.3(d)的Cytoscan图显示了基因组拷贝数图。图插图显示了使用Tilepath克隆以及映射BAC(C)和Fosmid(D)克隆的映射数据的荧光原位杂交(FISH)。请注意基于鱼图像的断点分配,描绘了14q11.2和19q13.3分别位于Tra@ dowr@下游增强子和下游短形式PVRL2的断点。差异信号强度符合焦点扩增,如两个基因座的拷贝数图所示。鱼类和基因组阵列。使用HISKY系统(Applied Spectral Imaging,Edingen,Germany)捕获了细胞遗传学图像,该系统配置为AxioImager D1 Micro-Scope(Zeiss,Jena,Germany)。如参考文献中所述,Siebert Lab友好地捐赠了克隆。10,或从美国加利福尼亚州奥克兰市的BACPAC资源,儿童医院购买,并由Nick Translation用Dutp Fluors Dy495(绿色),DY590(RED)和DY547(黄色)(黄色)(黄色)购买。基因组阵列数据由Cytoscan高密度基因组阵列(Affymetrix,Thermo Fischer,Darmstadt,Germany)提供。

NIPBL+
外染色体DNA(ECDNA)是癌症局灶性致癌基因扩增的中心机制,发生在大约15%的早期癌症和30%的晚期癌症中。ECDNA通过动态调节基因拷贝数并重新启动基因调节网络,驱动肿瘤形成,进化和耐药性。阐明ECDNA扩增的基因组结构对于理解肿瘤病理学和开发更有效的疗法至关重要。 配对的末端短阅读(Illumina)测序和映射已被用来使用断点图表示eCDNA扩增,其中将ECDNA的推断架构编码为图中的循环。 断点图的遍历已用于成功预测癌症样品中的ECDNA。 然而,在识别断点,复杂的重排和内部重复的鉴定以及ECDNA结构的细胞到细胞异质性的反卷积方面,短阅读技术在识别中固有限制。 长读技术,例如来自牛津纳米孔技术,具有改进推理的潜力,因为读取较长在映射结构变体方面更好,并且更有可能跨越重新排列或重复的区域。 在这里,我们提出了珊瑚(通过长读数的扩增完全重建),用于使用长阅读数据重建ecdna架构。 珊瑚重建可能使用二次编程的循环档案,同时优化重建的简约,解释了拷贝数和长阅读映射的一致性。 可用性:https://github.com/ampliconsuite/coral阐明ECDNA扩增的基因组结构对于理解肿瘤病理学和开发更有效的疗法至关重要。配对的末端短阅读(Illumina)测序和映射已被用来使用断点图表示eCDNA扩增,其中将ECDNA的推断架构编码为图中的循环。断点图的遍历已用于成功预测癌症样品中的ECDNA。然而,在识别断点,复杂的重排和内部重复的鉴定以及ECDNA结构的细胞到细胞异质性的反卷积方面,短阅读技术在识别中固有限制。长读技术,例如来自牛津纳米孔技术,具有改进推理的潜力,因为读取较长在映射结构变体方面更好,并且更有可能跨越重新排列或重复的区域。在这里,我们提出了珊瑚(通过长读数的扩增完全重建),用于使用长阅读数据重建ecdna架构。珊瑚重建可能使用二次编程的循环档案,同时优化重建的简约,解释了拷贝数和长阅读映射的一致性。可用性:https://github.com/ampliconsuite/coral与以前的基于简读的工具相比,珊瑚在广泛的模拟和9个数据集中的重建基本上改善了重建。随着长阅读的用法变得广泛,我们预计珊瑚将成为培养肿瘤中局灶性扩增的景观和演变的宝贵工具。


有机偏光发光二极管具有较高的亮度和颜色纯度朝向激光显示
抽象的宪法复杂染色体重排(CCR)是通过未知机制在种系中产生的罕见细胞遗传畸变。在这里,我们使用全面的基因组和表观基因组分析分析了微观三向或更复杂的易位的断点连接。所有这些易位连接均显示出伪造的基因组复杂性。这些断点聚集在小基因组域中,该结构域显示了微学或微插入。值得注意的是,所有从头案件都是父亲的起源。突破点分布特别对应于ATAC-SEQ(带测序的转座酶可访问染色质的测定)读取成熟精子的数据峰,而不是其他染色质标记或组织。我们提出,在脂肪生成后的精子发生过程中,CCR中的DNA断裂可能会在可接收的密集染色质区域中发展。