XiaoMi-AI文件搜索系统
World File Search System病例

附录1 - 病例定义和疾病
*对于在过去5-42天内用麻疹疫苗免疫的可疑麻疹的个体,需要麻疹病毒基因分型才能区分野生型与疫苗相关的麻疹。基因分型需要收集NAAT(PCR)PHO实验室标本,将在所有阳性样品上实施麻疹疫苗基因型PCR,以区分疫苗菌株。与疫苗相关的麻疹疾病(基因型A)不可报告,应报告为免疫后的不良事件(AEFI)。


创伤性脑损伤病例研究
数据集3:高中足球运动员认知,记忆和与脑震荡相关的问题是头部受伤的症状,但并非所有受伤都会导致脑震荡。在2018年发表的一项研究中,BU研究人员对8名青少年和年轻运动员的大脑进行了验尸分析。四人遭受了最近与运动有关的头部撞击伤害(死亡前1天到4个月),而其他四人没有头部撞击损伤的史。例如,案例3是一名17岁的男性高中美式足球和曲棍球运动员。他在生命中被诊断出患有两次与运动相关的脑震荡,这是死亡前2天的最后持续。案例8是一名22岁的男性前高中美式足球运动员。他遭受了三次脑震荡,所有脑震荡都在死亡前7年持续。

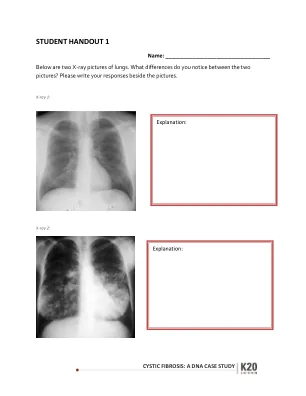
囊性纤维化:DNA病例研究
患有囊性纤维化的人在称为CFTR(囊性纤维化跨膜电导调节剂)的7号染色体上继承了一个缺陷的基因。该基因产生的蛋白质通常有助于盐(氯化钠)进出细胞。如果蛋白质无法正常工作,则该运动被阻塞,并且在细胞的外部产生异常厚的粘性粘液。最严重影响的细胞是肺部细胞。这种粘液堵塞肺部的气道,并增加了细菌感染的风险。

相关肺动脉高压:病例系列和
摘要 目的 本研究旨在通过病例系列和文献复习探讨妊娠合并系统性红斑狼疮相关肺动脉高压 (SLE-PAH) 的临床特点和结局。方法 本研究为单中心回顾性研究,纳入 2009 年至 2020 年期间北京协和医院经右心导管检查 (RHC) 确诊的 10 例连续妊娠合并 SLE-PAH 患者。查阅文献并回顾性分析 14 例妊娠合并 SLE-PAH 病例。结果 10 例患者初次就诊时平均年龄为 30.00±5.72 岁,SLE 和 PAH 的中位病程分别为 34.5(范围 1~164)个月和 2(1~51)个月。2 例患者有计划妊娠,7 例患者妊娠期间发生 PAH,1 例为非计划妊娠。另外,9 名患者为低度疾病活动性患者,系统性红斑狼疮疾病活动指数在 0 至 4 之间,30%、30% 和 40% 的患者分别属于 WHO 功能 II、III 和 IV 级。所有患者均接受了 RHC 和超声心动图检查。70% 的患者 N 端脑钠肽前体 (NT-proBNP) 水平升高,中位水平为 776(56-18 023)pg/mL。所有患者完成妊娠的中位时间为 31(15-38)周,6 名患者产下活婴。70% 的患者在产后 6 个月内 SLE 活动性和 PAH 严重程度得到改善。1 名患者在引产后第 15 天死亡。其余患者均达到狼疮低度疾病活动性状态;按照欧洲心脏病学会/欧洲呼吸学会风险分层,与妊娠期间的风险分层相比,7 例被归类为较低风险状态,2 例仍处于中等风险。此外,80% 的患者表现出轻度损害,WHO 功能分级为 I 或 II 级。产后 6 个月内 NT-proBNP 水平的中位数为 184(32~4003)pg/mL。在所回顾的文献中,患者平均年龄为 30.09±5.37 岁。完成妊娠的中位数为 36(28~40)周。更多病例有计划且成功妊娠,母亲和新生儿的存活率分别为 85.71% 和 92.86%。结论 如果在正确的治疗、严密的监测和全面的评估下达到 SLE-PAH 的治疗目标,SLE-PAH 女性是有可能成功怀孕的。

缺血性心脏病的临床病例
背景:自发性冠状动脉解剖(SCAD)是急性冠状动脉综合征的罕见原因,主要影响没有典型危险因素的年轻女性。由于其多样化的血管造影外观以及有组织的血栓等模仿的潜力,它带来了诊断挑战。我们报告了两名最初被诊断为SCAD的中年男性,后来被确认已组织了血栓。案例:两名中年男性,两个吸烟者,分别出现胸痛超过12个小时。均显示出ECG和前壁性低下的前铅的ST段升高,2D回波的LVEF为40%。紧急冠状动脉造影术显示与LAD中的多个辐射透明流明有关对比染料染色,表明潜在的SCAD(图1A和1C)。决策 - 做出:鉴于SCAD的这种异常出现,我们使用光学相干断层扫描(OCT)进行了进一步研究,该光学相干断层扫描(OCT)揭示了一个有组织的血栓,具有“瑞士奶酪”外观,与SCAD中的典型辐射式假腔形成对比(图1B和1D)。这两种情况都进行了成功的经皮冠状动脉干预,并进行了血栓抽吸和支架放置。


构造障碍的临床病例
‘我需要做什么才能有所作为?作为临床医生,可能是我们最重要的问题,经常被问到的问题。在这种有见地且易于访问的逻辑症状病例中,著名作者和编辑玛格丽特·沃尔什(Margaret Walshe)和尼克·米勒(Nick Miller)与受邀作者一起描述了许多相关且有益的案例示例,这些案例,这些案例揭示了与异性疾病和临床医生有关的交流问题所面临的交流问题的深度和复杂性。,他们热情地做到这一点。Walshe和Miller为我们提供了向个人学习的机会,从而在其他情况下找到适用的原则。我们获得了新的观点,而不仅仅是解决问题”。瑞典哥德堡大学卫生与康复系Lena Hartelius教授

病例课 - 神经外科杂志
缩写acoma =前交流动脉; Afr =孔圆形的动脉; apha =上升咽动脉; BMS =裸机支架; BTO =气球测试阻塞; CCA =海绵状颈动脉瘤; DAPT =双重抗血小板治疗; des =洗脱支架; DSA =数字减法血管造影; EC =颅外; ECA =外部颈动脉; ic =颅内; ICA =内部颈动脉; MRI =磁共振成像; NBCA = N-丁基-2-丙烯丙烯酸酯; PCI =经皮冠状动脉干预; pcoma =后验交流动脉; SPECT =单光子发射计算机断层扫描; TAE =经导管动脉栓塞; Vag =椎动脉血管造影。包括2025年2月24日发表的援引; doi:10.3171/case2469 3。于2024年10月7日提交。接受于2024年12月3日。