XiaoMi-AI文件搜索系统
World File Search SystemCRRNA

斑马鱼胚胎显微注射 - NET
此方案是使用已停产的 Cas9 蛋白版本 (Alt-R Sp Cas9 Nuclease 3NLS) 开发的。目前可用的产品 (Alt-R Cas9 Nuclease V3) 具有改进的 NLS,应以相同的体积和浓度直接替换到此方案中。IDT 建议使用 Alt-R™ Sp Cas9 Nuclease V3 与 Alt-R CRISPR-Cas9 crRNA 和 tracrRNA 结合使用,以生成核糖核蛋白编辑复合物,从而在大多数目标位点上实现高编辑效率。查看 Alt-R CRISPR-Cas9 用户指南,了解如何将核糖核蛋白转染哺乳动物细胞系(可在 www.idtdna.com/CRISPR 上找到)。
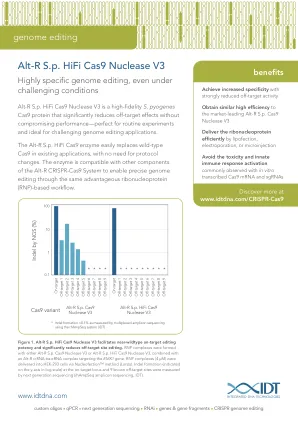
IDT Alt-R Sp HiFi Cas9 核酸酶 V3 (CRS-10083-FL 03/19)
图 1. Alt-R Sp HiFi Cas9 Nuclease V3 促进近野生型靶向编辑效力并显著减少脱靶位点编辑。RNP 复合物由 Alt-R Sp Cas9 Nuclease V3 或 Alt-R Sp HiFi Cas9 Nuclease V3 与靶向 EMX1 基因的 Alt-R crRNA:tracrRNA 复合物结合形成。RNP 复合物 (4 µM) 通过 Nucleofection™ 方法 (Lonza) 递送到 HEK-293 细胞中。通过下一代测序 (rhAmpSeq 扩增子测序,IDT) 测量了靶位点和 9 个已知脱靶位点处的插入/缺失形成 (在 y 轴上以对数标度表示)。

基于CRISPR-Cas的工具的开发和应用
成簇的规律间隔的短回文重复序列-CRISPR相关(CRISPR-Cas)系统作为细菌和古菌中一种重要的RNA引导的适应性免疫系统,其功能是防御病毒、质粒和转座子等移动遗传元件(MGEs)的侵害(Sorek et al., 2013; Faure et al., 2019; Koonin and Makarova, 2019; Makarova et al., 2019)。CRISPR位点由Cas基因和CRISPR阵列组成。CRISPR-Cas系统的功能主要分为三个阶段。第一阶段是适应阶段,Cas蛋白如Cas1和Cas2将外来的原型间隔序列插入到CRISPR阵列中,使其成为新的间隔物。第二阶段为表达阶段,CRISPR阵列转录为前CRISPR RNA(crRNA),随后加工为成熟的crRNA。最后是干扰阶段,crRNA引导CRISPR效应蛋白裂解病毒、质粒等外来靶序列(Barrangou et al., 2007; Brouns et al., 2008)。此前人们认为CRISPR系统仅存在于细菌和古菌中,但最近在巨型噬菌体中发现,CRISPR系统缺少适应阶段所需的Cas蛋白,如Cas1、Cas2和Cas4,而相应的效应蛋白也具备基因编辑能力(Al-Shayeb et al., 2020; Pausch et al., 2020)。这些CRISPR-Cas系统可能靶向宿主基因组,调控宿主基因表达,增强噬菌体的生存力(Al-Shayeb et al.,2020)。CRISPR-Cas系统与MGEs竞争,促进了CRISPR-Cas系统的进化,大大增加了其多样性(Koonin and Makarova,2019)。目前的CRISPR-Cas系统根据效应模块分为1类和2类(Makarova et al.,2015)。1类系统具有由多个Cas蛋白组成的效应模块,包括3种类型和16种亚型,而2类系统包含一个大蛋白,包括3种类型和17种亚型(Makarova et al.,2019)。在过去的十年中,CRISPR-Cas系统已经发展成为多种编辑工具。由于1类成员的复杂性,目前开发的基因编辑工具较少(Özcan等人,2021;Dolan等人,2019;Cameron等人,2019)。目前,2类成员正在被开发成大量的基因编辑工具。2类系统分为三类,包括II型、V型和


crispr/cas9介导的基因编辑
简介评论:引言有效地介绍了CRISPR/CAS9系统的历史背景,从而将其从Escherichia Coli的发现成为其作为基因编辑工具的发展。Jennifer Doudna和Emmanuelle Charpentier的贡献得到了充分的详细说明,强调了他们独立的研究工作和最终的合作。CRISPR机制的解释是彻底的,涵盖了关键组成部分,例如Cas9蛋白,引导RNA,tracrrna和crrna。基因编辑过程的分步分解,包括DNA裂解,序列靶向和基因剪接,为理解系统的功能提供了强大的基础。提及PAM序列及其在特异性中的作用可确保在解释目标位点选择方面的清晰度。
Alt -R CRISPR系统 - NET
*我们保证,使用Alt-R-R Crispr-Cas9指南RNA(CRRNA:TRACRRNA DUPLEX或SGRNA)和Alt-R S.P.cas9核酸酶或Alt-R S.P.HIFI Cas9核酸酶。 编辑分析必须在DNA水平上,例如使用Alt-R基因组编辑检测试剂盒或DNA测序。 如果未在成功的指导RNA中观察到成功的编辑,而在适当的阳性控制成功的同时,将批准对先前设计的ALT-R CRISPR-CAS9指南RNA进行一次“无成本”替换。 此保证不会扩展到任何替代产品,或任何其他发生或附带费用或费用。HIFI Cas9核酸酶。编辑分析必须在DNA水平上,例如使用Alt-R基因组编辑检测试剂盒或DNA测序。如果未在成功的指导RNA中观察到成功的编辑,而在适当的阳性控制成功的同时,将批准对先前设计的ALT-R CRISPR-CAS9指南RNA进行一次“无成本”替换。此保证不会扩展到任何替代产品,或任何其他发生或附带费用或费用。

Alt-R CRISPR-CAS9系统: - 核糖核蛋白的输送...
*我们保证,使用Alt-R-R Crispr-Cas9指南RNA(CRRNA:TRACRRNA DUPLEX或SGRNA)和Alt-R S.P.cas9核酸酶或Alt-R S.P.HIFI Cas9核酸酶。 编辑分析必须在DNA水平上,例如使用Alt-R基因组编辑检测试剂盒或DNA测序。 如果未在成功的指导RNA中观察到成功的编辑,而在适当的阳性控制成功的同时,将批准对先前设计的ALT-R CRISPR-CAS9指南RNA进行一次“无成本”替换。 此保证不会扩展到任何替代产品,或任何其他发生或附带费用或费用。HIFI Cas9核酸酶。编辑分析必须在DNA水平上,例如使用Alt-R基因组编辑检测试剂盒或DNA测序。如果未在成功的指导RNA中观察到成功的编辑,而在适当的阳性控制成功的同时,将批准对先前设计的ALT-R CRISPR-CAS9指南RNA进行一次“无成本”替换。此保证不会扩展到任何替代产品,或任何其他发生或附带费用或费用。

针对功能基因组学的工程化 CRISPR-Cas12a 的高阶组合染色质扰动
多重遗传扰动对于测试编码或非编码遗传元件之间的功能相互作用至关重要。与 DNA 切割相比,使用 CRISPR 干扰 (CRISPRi) 抑制染色质形成可避免基因毒性,并且在混合检测中更有效地扰乱非编码调控元件。然而,目前的 CRISPRi 混合筛选方法通常仅限于每个细胞靶向 1-3 个基因组位点。为了开发一种在功能基因组学筛选中使用 CRISPRi 对基因组位点进行高阶 (> 3) 组合靶向的工具,我们设计了一种 Acidaminococcus Cas12a 变体——称为多重转录干扰 AsCas12a (multiAsCas12a)。 multiAsCas12a 在使用慢病毒转导传递的 CRISPR RNA(crRNA)高阶多路复用阵列进行组合 CRISPRi 靶向时,其表现明显优于最先进的 Cas12a 变体,

遗传学
Cas,CRISPR 相关;CRISPR,成簇的规律间隔的短回文重复序列;CRISPRa,CRISPR 介导的转录激活;CRISPRi,CRISPR 介导的转录抑制;crRNA,CRISPR RNA;crRNP,CRISPR 核糖核蛋白;dCas9,核酸酶失活 Cas9;DSB,双链断裂;dsDNA,双链 DNA;dsODN,双链寡脱氧核苷酸;gRNA,向导 RNA;H3K27ac,组蛋白 H3 赖氨酸 27 乙酰化;H3K4me1,组蛋白 H3 赖氨酸 4 单甲基化;LAM-PCR,线性扩增介导的 PCR;LSD1,赖氨酸特异性组蛋白去甲基化酶 1;MCP,MS2 外壳蛋白;MOI,感染复数; p65AD,核因子-κB反式激活亚基激活结构域;PAM,原型间隔区相邻基序;RNAi,RNA干扰;scFV,单链可变片段;sfGFP,超折叠GFP;sgRNA,单向导RNA;ssRNA,单链RNA。

针对目标基因组工程的紧凑型级联cas3系统
CRISPR-CAS技术提供了彻底改变研究的可编程基因编辑工具。领先的CRISPR-CAS9和CAS12A酶非常适合编程的基因操作,但是,它们受到基因组规模干预措施的限制。在这里,我们利用了一个基于CAS3的系统,该系统具有用于基因组工程的过程核酸酶。使用单个CRRNA编程的最小Cascade-CAS3系统(I型I-C)进行了优化,以产生效率接近100%的缺失,并用于迅速产生含量为7-424 kb的大删除,铜绿铜。相比之下,CAS9产生了小的缺失和点突变。CAS3生成的缺失边界是高度可变的,但通过同源指导修复(HDR)模板成功指定。HDR效率要高得多。最小I-C系统