XiaoMi-AI文件搜索系统
World File Search SystemFDASIA

向国会关于BPCA和PREA的儿科报告
2012年制定的《食品药品监督安全与创新法》(FDASIA)第508条要求卫生和公共服务部长报告有关第505A节和505B节的联邦食品,药物和化妆品和化妆品法案(FD&C ACT)的实施(FD&C ACT),通常被称为“最佳儿童ACTICE ACTICE ACTICA ACTICA and PEDICA”(BPCA),以及PEDICA ACTIACCA(BPCA)。该报告首先在2016年7月9日之前提交国会,然后每5年提交。除了FDASIA要求外,2017年的FDA重新授权法案(FDARA)还增加了对HHS的更多报告要求,包括与(1)(1)针对癌症治疗的儿科研究和标记有关的要求,以及(2)根据BPCA和PREA提出儿科研究的时间。根据FDASIA和FDARA提交的该报告提供了对BPCA和PREA的实施的评估,以及这些法规的影响,(2)突出了这两种法规所取得的其他成功,(3)提供了通过在BPCA和Prea中实施的目标,并有效地实施了精神药物,并有效地实施了有效的bpca和Prea效率。
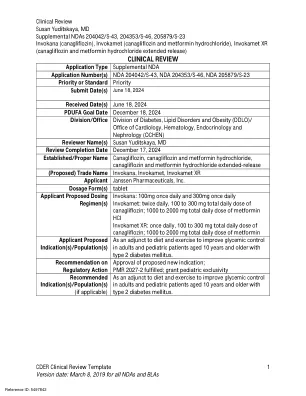
临床评论
AC advisory committee AE adverse event AESI adverse event of special interest AHA antihyperglycemic agent AR adverse reaction BLA biologics license application BPCA Best Pharmaceuticals for Children Act BRF Benefit Risk Framework CBER Center for Biologics Evaluation and Research CDER Center for Drug Evaluation and Research CDRH Center for Devices and Radiological Health CDTL Cross-Discipline Team Leader CFR Code of Federal Regulations CIs clinical investigator CI confidence interval CMC chemistry, manufacturing, and controls COSTART Coding Symbols for Thesaurus of Adverse Reaction Terms CRF case report form CRO contract research organization CRT clinical review template CSR clinical study report CVD cardiovascular death DBP diastolic blood pressure DMC data monitoring committee DKA diabetic ketoacidosis ECG electrocardiogram eCTD electronic common technical document eGFR estimated glomerular filtration速率ER扩展释放的ETASU元素可确保安全使用FAS完整分析集FDA食品药物管理局FDAAA食品和药物管理局修正案2007年FDASIA FDASIA食品和药物管理局安全和创新法FDCP固定剂量组合产品FPG禁食产品FPG禁食等质量等离子体葡萄糖

ONADE 政策和程序 1243.4068 包含支持安全性和有效性的外国数据提交的可接受性
《联邦食品药品和化妆品法案》(FD&C 法案)第 569B 条经 2012 年《食品药品管理局安全和创新法案》(FDASIA)修改,将 FDA 长期以来接受国外临床数据来支持申请的做法编入法典,前提是申请人证明数据“符合适用标准以支持批准”。CVM 致力于支持全球批准,以加强动物药物开发、促进国外数据的使用并尽量减少重复研究的需要。CVM 还支持国际协调活动,帮助利用其他专家机构的工作和专业知识。所有与安全性和有效性评估相关的国外数据都必须按照 21 CFR 514.1(b)(8)(iv) 的规定提交到新动物药物申请(NADA)、新动物药物有条件批准申请(CNADA)或试验性新动物药物(INAD)文件中。此要求也适用于简化的新动物药物申请 (ANADA) 和仿制药 INAD (JINAD) 文件。申请人必须提交美国境外的调查或商业营销数据(如果可以的话),无论这些数据是有利的还是不利的。

213535Orig1s000 临床评论 - accessdata.fda.gov
AC 咨询委员会 AE 不良事件 BLA 生物制品许可申请 BPCA 最佳儿童药物法案 BRF 效益风险框架 CBER 生物制品评价与研究中心 CCOD 临床截止日期 CDER 药品评价与研究中心 CDRH 设备和放射健康中心 CDTL 跨学科团队负责人 CFR 联邦法规 CHOP INTEND 费城儿童医院婴儿神经肌肉疾病检测 CMC 化学、制造和控制 COSTART 不良反应术语库的编码符号 CRF 病例报告表 CRO 合同研究组织 CRT 临床审查模板 CSF 脑脊液 CSR 临床研究报告 CSS 管制物质工作人员 DMC 数据监测委员会 ECG 心电图 eCTD 电子通用技术文档 ETASU 确保安全使用的要素 FDA 食品药品管理局 FDAAA 2007 年食品药品管理局修正案 FDASIA 食品药品管理局安全和创新法案 FEV1 1 秒用力呼气量 FVC 用力肺活量 GCP良好的临床实践 GRMP 良好的评审管理实践 HFMSE 汉默史密斯功能运动量表 - 扩展版 HINE 汉默史密斯婴儿神经系统检查 ICH 国际协调会议 iDMC 独立数据监察委员会 IMC 独立监察委员会 IND 研究性新药 ISE 疗效综合总结

2024年9月20日,Gressitec Suji Shetty高管副副副作用...
2012年7月9日,《食品和药物管理局安全与创新法》第607条(FDASIA)修改了《食品,药物和化妆品法》第513(f)(2)条。本法律提供了两个从头分类的选项。首先,对于响应510(k)的“基本上不相同”(NSE)的任何人,该设备先前尚未根据该法案进行分类的设备,都可以要求FDA根据该法案的第513(a)(1)条对该设备进行基于风险的分类。2016年12月13日,《 21世纪治疗法》删除了一项要求,要求在收到NSE确定后的30天内提交新的请求。另外,任何确定没有合法销售的设备来基于确定实质性等效性的任何人都可以要求FDA根据该法案第513(a)(1)条对该设备进行基于风险的分类,而不会先提交510(k)。FDA应在收到此请求后的120天内对设备进行分类。此分类应为设备的初始分类。在发布该设备的订单后的30天内,FDA必须在宣布分类的联邦注册簿中发布通知。2023年9月28日,FDA收到了您的从头要求对Chronos®的分类。该请求是根据《 FD&C法案》第513(f)(2)条提交的。为了将Chronos®分类为I类或II类,拟议类有足够的监管控制,以合理地保证设备的安全性和有效性以供其预期使用。在审查了从头请求中提交的信息后,FDA确定,对于先前指定的使用指示,可以将Chronos®分类为II类,并建立II类特殊控件。FDA认为,II类(特殊)控件可以合理保证设备类型的安全性和有效性。下表总结了与设备类型相关的确定风险和缓解措施:

DEN230090.pdf -AccessData.fda.gov
测试检测与外行用户执行的与梅毒相关的抗体的检测。测试检测与外行用户执行的与梅毒相关的抗体的检测是一种体外诊断装置,用于检测用于家庭设置或类似环境的临床样本中的抗体。该设备旨在帮助诊断梅毒,旨在用于处方或非处方使用。2012年7月9日,《食品和药物管理局安全与创新法》第607条(FDASIA)修改了《食品,药物和化妆品法》第513(f)(2)条。本法律提供了两个从头分类的选项。首先,对于响应510(k)的“基本上不相同”(NSE)的任何人,该设备先前尚未根据该法案进行分类的设备,都可以要求FDA根据该法案的第513(a)(1)条对该设备进行基于风险的分类。2016年12月13日,《 21世纪治疗法》删除了一项要求,要求在收到NSE确定后的30天内提交新的请求。另外,任何确定没有合法销售的设备来基于确定实质性等效性的任何人都可以要求FDA根据该法案第513(a)(1)条对该设备进行基于风险的分类,而不会先提交510(k)。FDA应在收到此请求后的120天内对设备进行分类。此分类应为设备的初始分类。2023年12月27日,FDA收到了您的从头开始,要求对梅毒测试的第一个分类。在发布该设备的订单后的30天内,FDA必须在宣布分类的联邦注册簿中发布通知。该请求是根据《 FD&C法案》第513(f)(2)条提交的。为了将第一个了解梅毒测试的人分类为I类或II类,拟议类有足够的监管控制,以合理地保证该设备的安全性和有效性以供其预期使用。在审查了从头请求中提交的信息后,FDA确定,对于先前指定的使用指示,第一个知道梅毒测试的疾病可以在II类中归类为II类特殊控件。FDA认为,II类(特殊)控件可以合理保证设备类型的安全性和有效性。下表总结了与设备类型相关的确定风险和缓解措施:

指导文件
作者注 这项工作始于 2017 年 5 月,在 MRCT 中心生物伦理学协作会上,这是一个中立的论坛,许多临床研究利益相关者在此召开会议,讨论临床试验的多样性问题。会议的与会者由来自业界、学术界、政府和患者权益保护等多方利益相关者组成,他们一致认为,临床试验的参与者群体至少应该代表一般人群,最好能代表干预的目标人群。如果研究人群存在偏差,缺乏多样性,那么就无法充分研究和理解医疗干预的安全性和有效性、效果和价值——治疗效果的生物异质性。公正问题也会影响研究人群的多样性(或缺乏多样性),如果特定人群承担不成比例的负担或被不公平地排除在研究之外,就会被视为根本不公平。与会者一致认为,尽管从科学和伦理角度看,性别、性取向、族裔和种族少数群体在药物开发和临床研究中的代表性不足问题仍然存在。2015 年 1 月,FDA 首次发布《药物试验快照》,突显了这一问题的严重程度,尤其是对于代表性不足和服务不足的人群而言。1《药物试验快照》报告了参与当年导致产品获批的新分子实体 (NME) 或生物制品许可申请 (BLA) 关键试验的患者的人口统计学特征(性别、年龄、种族、民族)。该报告是真正的“快照”,取决于一个监管机构在一年内批准的药物和生物制品的变化情况。由于这一限制,药品评估和研究中心发布了一份出版物,该出版物部分是为了响应 2012 年食品药品管理局安全和创新法案 (FDASIA 907) 而发布的,揭示了性别和种族参与方面存在显著差异。2015 年,在批准的 45 种新药中,有超过 105,000 名参与者参与,但只有 40% 的患者是女性,而令人惊讶的是,只有 5% 是非裔美国人。然而,在 2015 年和 2016 年的两年时间内,有 67 种产品获得批准,但治疗领域存在巨大差异:在心血管疾病(2.50%)和肿瘤学(2.74%)产品试验中,黑人或非裔美国人患者所占比例不到总数的 3%,而在精神障碍试验中 24.18% 的参与者是黑人或非裔美国人。2 因此,临床试验参与和药物开发的种族多样性是可能的,只是没有发生,而且显然没有被优先考虑。