XiaoMi-AI文件搜索系统
World File Search SystemKras

KRAS Q61H 突变使癌细胞获得对 SHP2 抑制的耐药性
携带不同 KRAS 突变的癌细胞对 SHP2 抑制的敏感性也不同。《自然通讯》最近发表的一项研究揭示了携带 KRAS Q61H 突变的癌细胞对 SHP2 抑制剂(SHP2i)的潜在耐药机制。1这项研究表明,KRAS Q61H 突变通过将 KRAS 与 SHP2 介导的上游核苷酸交换因子(鸟嘌呤核苷酸交换因子 [GEF])/GTPase 活化蛋白 (GAP) 调控分离而使癌细胞对 SHP2i 产生耐药性,为治疗携带 KRAS Q61H 突变的癌症提供了新的见解。KRAS 是突变最常见的 RAS 亚型,是一种编码小 GTPase 转导蛋白的原癌基因。响应上游信号,KRAS 可以通过 GEF(例如 Son of Sevenless (SOS) 或 GAP)在无活性的二磷酸鸟苷 (GDP) 状态和活性的三磷酸鸟苷 (GTP) 状态之间切换。2 KRAS 突变主要发生在密码子 12、13 或 61,占 RAS 突变的 86%。特别是,谷氨酰胺 61 通过定位攻击水分子和稳定水解反应的过渡态,在催化过程中起直接作用。3,4 通常,突变的 KRAS 可通过影响 GAP 介导的 GTP 水解导致活性 GTP 结合的 KRAS 积累,从而导致 RAS – RAF – MEK – ERK 通路过度活化,并伴有不受控制的细胞增殖。4 KRAS 突变在许多人类癌症中很常见,尤其是胰腺癌、非小细胞肺癌和结直肠癌。值得一提的是,特定的 KRAS 突变可能导致肿瘤患者的不同预后和治疗反应。因此,KRAS 突变对癌症治疗研究人员提出了挑战。2,4,5 从历史上看,KRAS 一直被认为是“不可成药”的药物靶点,因为它不包含经典的可用于药物的小分子结合口袋。6 通过关闭致癌基因,已经开发出用于抗癌药物开发的间接和直接方法

Igf2bp1 的小分子抑制剂可抑制癌细胞中的 Kras 和促癌表型
摘要 Igf2bp1 是一种癌胚 RNA 结合蛋白,其在多种类型的癌症中的表达与关键促癌 RNA 的上调、预后不良和生存率降低有关。重要的是,Igf2bp1 与 Kras 突变协同作用,增强信号传导和致癌活性,这表明抑制 Igf2bp1 的分子可能具有治疗潜力。在这里,我们分离出一种小分子,它与 Igf2bp1 KH3 和 KH4 结构域边界的疏水表面相互作用,并抑制与 Kras RNA 的结合。在细胞中,该化合物降低 Kras 和其他 Igf2bp1 mRNA 靶标的水平,降低 Kras 蛋白,并抑制下游信号传导、伤口愈合和软琼脂中的生长,所有这些都没有任何毒性。这项研究为改善肺癌和其他癌症中表达 Igf2bp1 的肿瘤的预后提供了一种途径。

致癌基因突变或致癌基因 KRAS 或 BRAF 的过度表达不足以导致致癌基因成瘾
。CC-BY-NC-ND 4.0 国际许可,根据 未经同行评审认证)是作者/资助者,他已授予 bioRxiv 永久展示预印本的许可。它是此预印本的版权持有者(此版本于 2020 年 5 月 29 日发布。;https://doi.org/10.1101/2020.05.26.117911 doi:bioRxiv 预印本

针对 p53 和 KRAS 的基因疗法治疗结直肠癌:神话还是前进的方向?
摘要:结直肠癌 (CRC) 是全球第三大最常见的恶性肿瘤,是男性和女性死亡的主要原因之一。尽管人们付出了巨大的努力来提高公众对早期筛查的认识,并在 CRC 治疗方面取得了重大进展,但大多数病例仍然在晚期才被诊断出来。这导致这种癌症的存活率较低。CRC 患者表现出各种基因变化和表观遗传修饰。与 CRC 相关的最常见基因改变是 p53 和 KRAS 突变。针对 CRC 中的缺陷基因(如 TP53(编码 p53 的肿瘤抑制基因)和 KRAS(致癌基因))的基因治疗可能成为除标准疗法之外的另一种治疗途径。在过去十年中,用于治疗各种癌症的基因治疗取得了重大进展。这包括开发载体作为运载工具。尽管靶向基因治疗在癌症治疗中被看好,但它也存在各种局限性,例如缺乏相关技术、所涉及的程序成本高以及伦理问题。本文将对针对 p53 和 KRAS 的基因治疗在 CRC 治疗中的潜力和挑战进行综述。

FGFR1 和 PLK1 抑制剂的协同作用靶向 KRAS 突变癌症的代谢缺陷
KRAS 癌蛋白在人类癌症中常见突变,但专门针对 KRAS 驱动肿瘤的有效疗法仍然难以捉摸。在这里,我们表明成纤维细胞生长因子受体 1(FGFR 1)和 polo 样激酶 1(PLK 1)抑制剂联合治疗在体外和体内 KRAS 突变肿瘤模型中引起协同细胞毒性。FGFR 1 和 PLK 1 的药理学和遗传抑制产生协同作用,增强 KRAS 突变肺癌和胰腺癌(但不影响结肠癌和 KRAS 野生型癌细胞)的抗增殖作用和细胞死亡。从机制上讲,共同靶向 FGFR 1 和 PLK 1 会上调活性氧(ROS),导致氧化应激激活的 c-Jun N 末端激酶(JNK)/p 38 通路和 E 2 F 1 诱导的细胞凋亡。我们进一步阐明了自噬可防止 PLK 1 /FGFR 1 抑制剂的细胞毒性,而通过临床批准的氯喹拮抗补偿机制可充分发挥 PLK 1 和 FGFR 1 靶向治疗的治疗潜力,在 KRAS 突变患者来源的异种移植和 Kras 诱发的肺腺癌基因工程小鼠模型中产生强效且持久的反应。这些结果表明 FGFR 1 和 PLK 1 在代谢应激监测中发挥着以前未被重视的作用,并展示了一种用于治疗 KRAS 突变癌症的协同药物组合。

通过天然产物的低毒性化合物突变的KRAS癌球体的生长抑制
摘要。背景/目标:在天然产物的化合物中,选择性抑制了具有突变体(MT)Kras,NP910的癌症生长的化合物,并探索了其衍生物。材料和方法:表达野生型(WT)KRAS(HKE3-WTKRAS)和MTKRAS(HKE3-MTKRA)的HKE3球体面积在三维浮动(3DF)培养物中测量,并用18 NP910衍生物处理。通过长期3DF(LT3DF)培养和裸小鼠测定法测定50%细胞生长抑制(GI50)。结果:我们选择了NP882(命名为STAR3)作为HKE3-MTKRAS球体生长的最有效抑制剂,NP910衍生物中毒性最少。Gi50s和裸鼠测定法分别为6μm和30.75 mg/kg。然而,在50%的细胞系中观察到与KRAS突变无关的细胞系抑制,这表明Star3的靶标与KRAS突变和KRAS相关信号没有直接相关。结论:STAR3是一种低毒性化合物,可抑制某些肿瘤细胞的生长。

改进的PDE6D抑制剂与西地那非结合以抑制KRAS突变癌细胞生长
摘要:提出了贩运伴侣PDE6D(或PDEδ)作为K-RAS的替代靶标,导致一系列阻断其前蛋白酶结合口袋的抑制剂的发展。这些抑制剂的溶解度低和可疑的脱靶效应,从而阻止了它们的临床发育。在这里,我们开发了一种高度可溶的纳摩尔PDE6D抑制剂(PDE6DI),Deltaflexin3,其具有最低的脱靶活性,与三种突出的参考化合物相比。deltaflexin3降低了RAS信号传导,并有选择地降低了KRAS突变体和PDE6D依赖性癌细胞的生长。我们进一步表明,pKG2介导的Ser181的磷酸化降低了与PDE6D结合的K-RAS。因此,Deltaflexin3与认可的PKG2激活剂西地那非结合使用,以更有效地抑制PDE6D/K-RAS结合,癌细胞增殖和微肿瘤生长。如前所述,RAS运输,信号传导和癌细胞增殖的抑制作用仍然适中。我们的结果表明将PDE6D重新评估为癌症中的K-Ras替代靶标。■简介

Vegen Labs获得3.76 cr的BIRAC资金以推进KRAS抑制剂
肺癌仍然是印度与癌症相关死亡的主要原因之一,NSCLC约占所有肺癌病例的85%。,印度每年都会报告超过70,000例新的肺癌病例,许多患者在高级阶段被诊断为有针对性的疗法,例如KRAS抑制剂,可以提供改变生活的治疗方法。随着意识,早期诊断和获得先进疗法的访问,印度NSCLC市场有望实现显着增长,从而对创新的癌症治疗产生了强劲的需求。

针对非小细胞肺癌中驱动 KRAS 突变的有效药物组合
摘要:药物基因组学是一个快速发展的领域,其目标是为每位患者提供个性化治疗 1。此前,我们开发了新药机会计算分析 2 (CANDO) 平台,用于多尺度治疗发现,通过分析化合物与大型蛋白质库的相互作用,筛选出针对任何 3 适应症/疾病的最佳化合物。我们在 4 CANDO 5 平台内实施了全面的精准医疗药物发现流程,以确定哪些药物最有可能对非小细胞肺癌 (NSCLC) 的突变表型有效,其依据的假设是具有相似相互作用 7 特征(或特征)的药物将具有相似的行为,因此表现出协同作用。CANDO 8 预测 EGFR 抑制剂奥希替尼最有可能与四种 KRAS 抑制剂产生协同作用。 9 细胞增殖试验验证研究证实,奥希替尼与 10 ARS-1620(一种 KRAS G12C 抑制剂)和 BAY-293(一种泛 KRAS 抑制剂)联合使用,通过作用于突变型 KRAS 表现出协同作用,11 降低细胞增殖。我们的精准医疗管道可用于 12 识别能够与 KRAS G12C 抑制剂产生协同作用的化合物,并通过了解它们在蛋白质组学/相互作用组学尺度上的行为来评估它们 13 成为药物的可能性。14
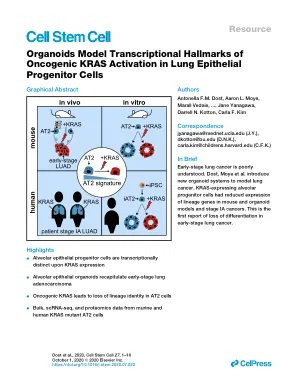
类器官模型肺上皮祖细胞中致癌 KRAS 激活的转录标志
Antonella FM Dost, 1 , 2 , 3 , 17 Aaron L. Moye, 1 , 2 , 3 , 17 Marall Vedaie, 4 , 5 Linh M. Tran, 6 Eileen Fung, 7 Dar Heinze, 4 , 8 Carlos Villacorta-Marting, 5 , 19 , Ryan Heman Julian H. Kwan, 9 , 10 Benjamin C. Blum, 9 , 10 Sharon M. Louie, 1 , 2 , 3 Samuel P. Rowbotham, 1 , 2 , 3 Julio Sainz de Aja, 1 , 2 , 3 Mary E. Piper, 11 Preetida J. Bhetariya , 1 , 1 , T Roderick . Bronson, 12 Andrew Emili, 9 , 10 , 13 Gustavo Mostoslavsky, 4 , 8 Gregory A. Fishbein, 14 William D. Wallace, 14 , 15 Kostyantyn Krysan, 6 Steven M. Dubinett, 6 , 16 Jane Yanaga , 17 , 4 , 4 * Darrell * N. , * and Carla F. Kim 1 , 2 , 3 , 18 , * 1 Stem Cell Program and Divisions of Hematology/Oncology and Pulmonary Medicine, Boston Children's Hospital, Boston, MA 02115, USA 2 Harvard Stem Cell Institute, Cambridge, MA 02133 Department of Genetics, Harvard Medical School, MA, Boston 5, USA 4 Center for Regenerative Medicine of Boston University and Boston Medical Center, Boston, MA 02118, USA 5 The Pulmonary Center and Department of Medicine, Boston University School of Medicine, Boston, MA 02118, USA 6 Department of Medicine, David Geffen School of Medicine at UCLA, University of Los Angeles, Los Angeles, CA, David Geffen School of Medicine, CA cine at UCLA, University of California, Los Angeles, Los Angeles, CA, USA 8 Section of Gastroenterology and Department of Medicine, Boston University School of Medicine, Boston, MA 02118, USA 9 Center for Network Systems Biology, Boston University, Boston, MA 02118, USA 10 Department of Biochemistry, Boston University School of Medicine, MA, MA of Public Health, Department of Biostatistics, Boston, MA 02115, USA 12 Rodent Histopathology Core, Harvard Medical School, Boston, MA 02115, USA 13 Department of Biology, Boston University, Boston, MA 02215, USA 14 Department of Pathology and Laboratory Medicine, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA 15 Department of Pathology, Keck School of Medicine of USC, University of Southern California, Los Angeles, CA 90033, USA 16 Jonsson Comprehensive Cancer Center, University of California, Los Angeles, Los Angeles, CA 90095, USA 17 These authors contributed equally 18. Contact: Contact Letters: Connected with Legal. JY), dkotton@bu.edu (DNK), carla.kim@childrens.harvard.edu (CFK) https://doi.org/10.1016/j.stem.2020.07.022