XiaoMi-AI文件搜索系统
World File Search SystemTNG908

stearoyl-COA去饱和酶是SMAD4缺陷癌症中的合成致命靶标
与功能基因组学研究结合的大规模DNA测序在表征癌症基因组方面起着关键作用,揭示了缺失事件的重要性,这些事件的重要性通过肿瘤抑制基因的丧失来促进肿瘤生长。诸如癌症基因组图集计划(TCGA)之类的倡议提供了整个人类癌症遗传改变的综合图,表明缺失事件通常延伸到肿瘤抑制基因基因座,从而导致相邻基因的代码。尽管这些乘客事件可能不会赋予肿瘤的直接健身优势,但它们可以创建可以通过治疗剥削的副脆弱性。一个例子是由甲基腺苷磷酸化酶(MTAP)丧失赋予PRMT5抑制作用的附带脆弱性,该基因经常与描述良好的肿瘤抑制基因CDKN2A相关。1-3 MTAP编码蛋白质MTAP,蛋白MTAP是蛋氨酸拯救途径中的临界酶,该过程从多胺合成的副产物中循环蛋氨酸,甲基噻吩腺苷(MTA)。CDKN2A的丧失发生在所有人类癌症中的10-15%中,并且在组织学上的频率更高,例如恶性周围神经鞘肿瘤,胶质母细胞瘤(GBM),间皮瘤,间皮瘤,尿路上皮癌,食管鳞状细胞癌,胰腺癌,胰腺腺瘤腺瘤,<- <- <-

TNG908,一种脑渗透剂MTA合作PRMT5抑制剂,在临床前胶质母细胞瘤模型中有效
Minjie Zhang*,Alice Tsai*,Kevin M Cottrell,Brian B Haines,Erik Wilker,Heather Dibenedetto,Ron Weitzman,Alan Huang,Charles B Davis,John P Davis,John P Maxwell和Kimberly J Briggs*这些作者均等贡献

发现TNG908:选择性,大脑渗透物,MTAMTA合件PRMT5抑制剂在MTAP中有效...MTA合件PRMT5抑制剂在MTAP中有效...
目标:恶性外周神经鞘肿瘤(MPNST)是高度侵略性的麦芽瘤,治疗率有限,存活率较差,因此需要发展新的治疗疗法。由于由近端抑制基因CDKN2A损失造成的乘客缺失,大约25-50%的MPNST港口丧失酶甲基腺苷磷酸化酶(MTAP)损失。PRMT5由于底物甲基噻吩并腺苷(MTA)的积累而被鉴定为MTAP被骨化细胞中的选择性依赖性,该细胞本身就是内源性PRMT5抑制剂。TNG908和TNG462是临床阶段MTA合件PRMT5抑制剂,可分别证明对MTAP细胞的选择性分别在15倍和45倍的MTAP -INTACT -INTACT细胞上。先前的报道表明,这两个分子都驱动了各种MTAP癌症组织学的异种移植模型中耐用的肿瘤回归。在这里,我们的目标是检查临床前MPNST模型中TNG908和TNG462的活性。
Adam Yaari
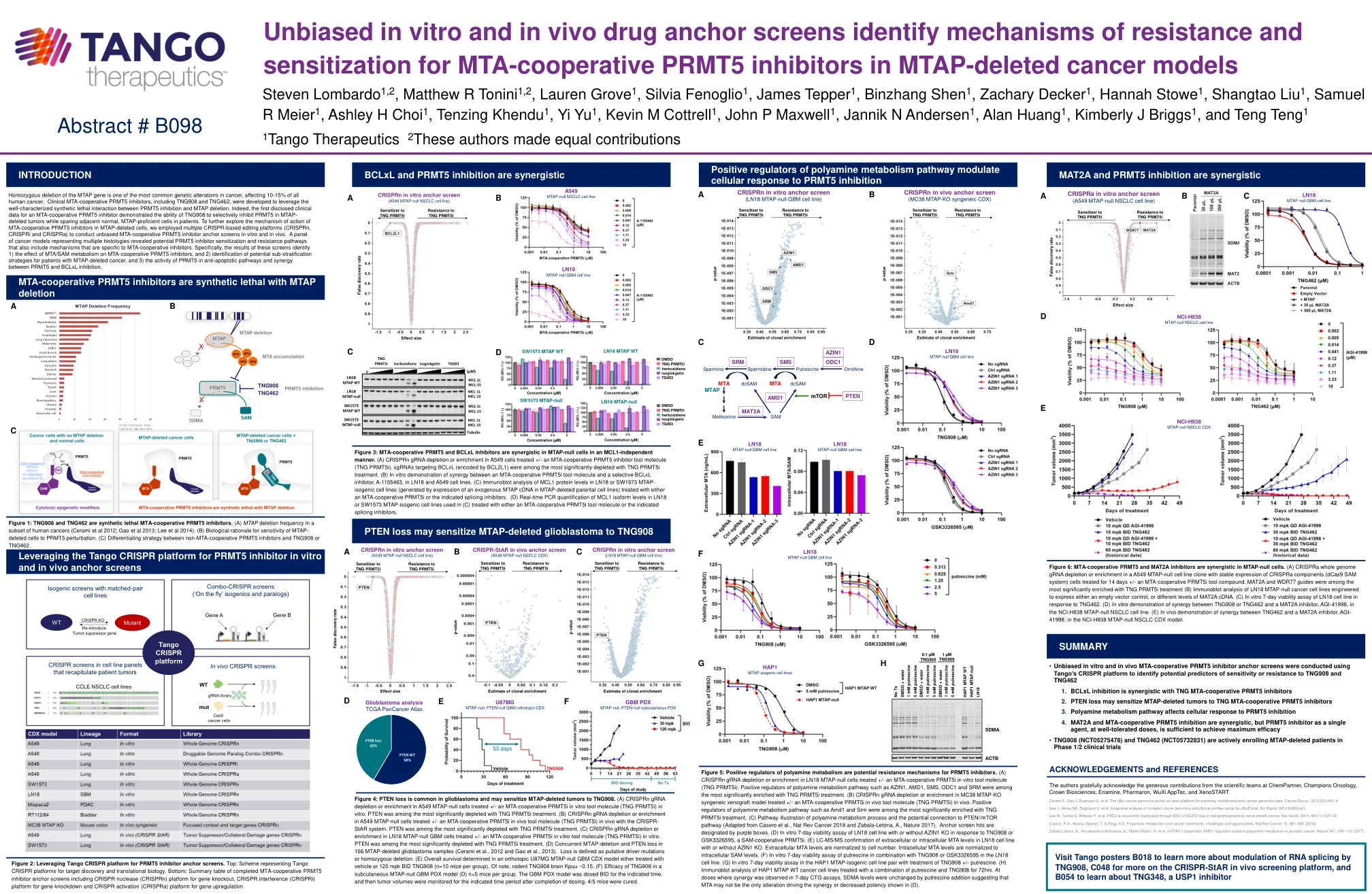
MTAP基因的纯合缺失是癌症中最常见的遗传改变之一,影响了所有人类癌症的10-15%。开发了包括TNG908和TNG462在内的临床MTA合并PRMT5抑制剂,以利用PRMT5抑制和MTAP缺失之间充分表征的合成致死相互作用。的确,第一个披露的MTA合作性PRMT5抑制剂的临床数据证明了TNG908在MTAP删除的肿瘤中有选择性地抑制PRMT5的能力,同时在患者中保留邻近的正常,MTAP-Profofoffic-Profofoffic-MTAP-Profofoffoffient。为了进一步探索MTA骨骼删除细胞中MTA合件性PRMT5抑制剂的作用机理,我们采用了多个基于CRISPR的编辑平台(CRISPRN,CRISPRI和CRISPRA)来进行无偏MTA合件性PRMT5抑制剂锚固筛网,并在In In In In In Intro和Vivo中进行。代表多种组织学的癌症模型揭示了潜在的PRMT5抑制剂敏化和耐药途径,这些途径还包括特定于MTA合并抑制剂的机制。Specifically, the results of these screens identify 1) the effect of MTA/SAM metabolism on MTA-cooperative PRMT5 inhibitors, and 2) identification of potential sub-stratification strategies for patients with MTAP-deleted cancer, and 3) the activity of PRMT5 in anti-apoptotic pathways and synergy between PRMT5 and BCLxL inhibition.
TNG462 是一种潜在的同类最佳 MTA 协同 PRMT5 抑制剂,可用于治疗 MTAP 缺失的实体瘤
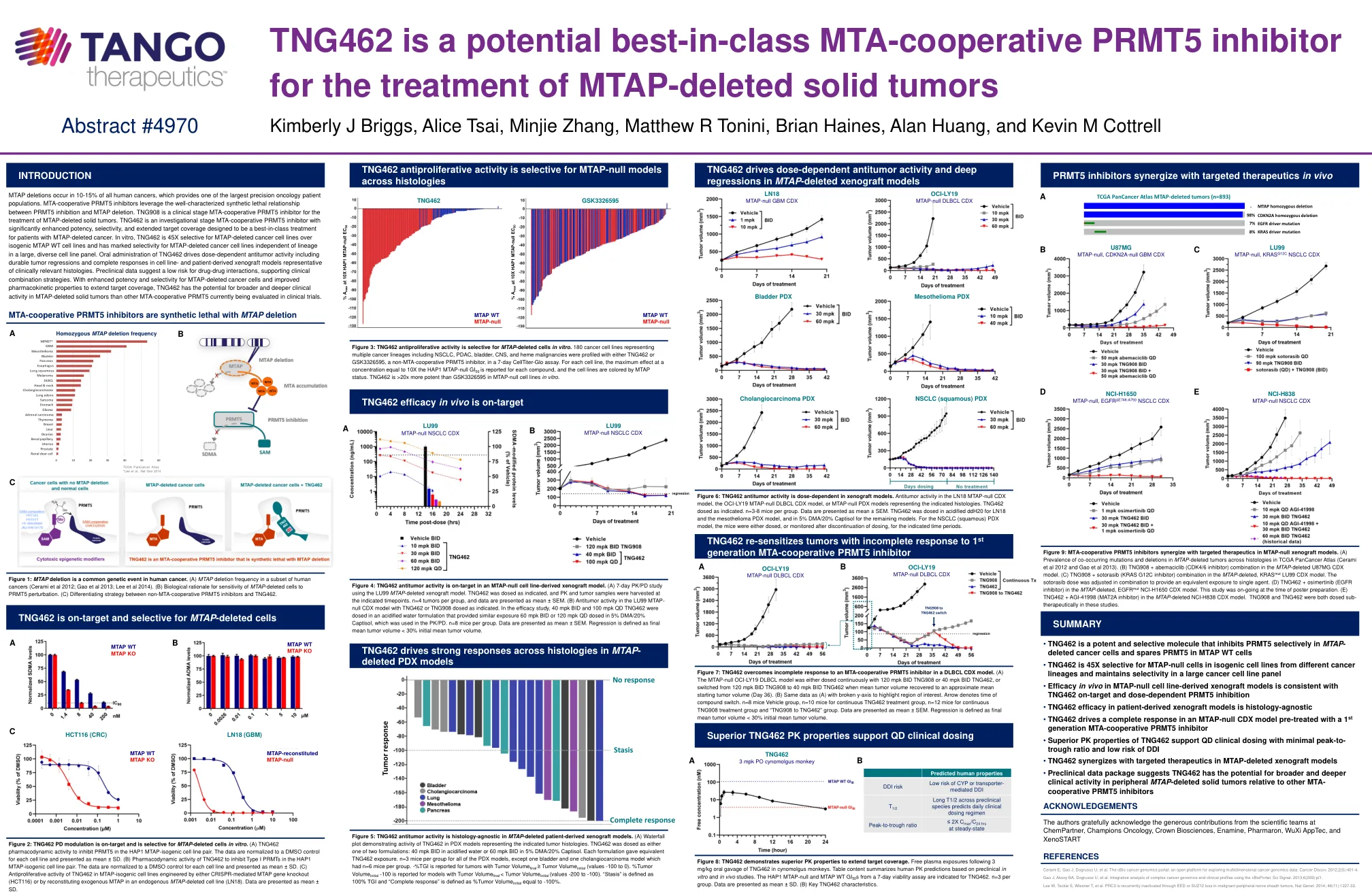
MTAP 缺失发生在 10-15% 的人类癌症中,这提供了最大的精准肿瘤患者群体之一。MTA 合作 PRMT5 抑制剂利用 PRMT5 抑制和 MTAP 缺失之间已充分表征的合成致死关系。TNG908 是一种临床阶段的 MTA 合作 PRMT5 抑制剂,用于治疗 MTAP 缺失的实体瘤。TNG462 是一种研究阶段的 MTA 合作 PRMT5 抑制剂,具有显著增强的效力、选择性和扩大的靶标覆盖范围,旨在成为 MTAP 缺失癌症患者的最佳治疗方案。在体外,TNG462 对 MTAP 缺失癌细胞系的选择性是同源 MTAP WT 细胞系的 45 倍,并且在大型多样化细胞系组中对 MTAP 缺失癌细胞系具有显着的选择性,与谱系无关。口服 TNG462 可产生剂量依赖性抗肿瘤活性,包括持久的肿瘤消退和代表临床相关组织学的细胞系和患者衍生异种移植模型中的完全反应。临床前数据表明药物相互作用风险低,支持临床联合策略。由于对 MTAP 缺失癌细胞的效力和选择性增强,以及药代动力学特性改善以扩大靶标覆盖范围,TNG462 有可能在 MTAP 缺失实体瘤中发挥比目前正在临床试验中评估的其他 MTA 合作 PRMT5 更广泛和更深的临床活性。
评估纯合MTAP截断对MTA合作PRMT5抑制剂的活性和选择性的影响
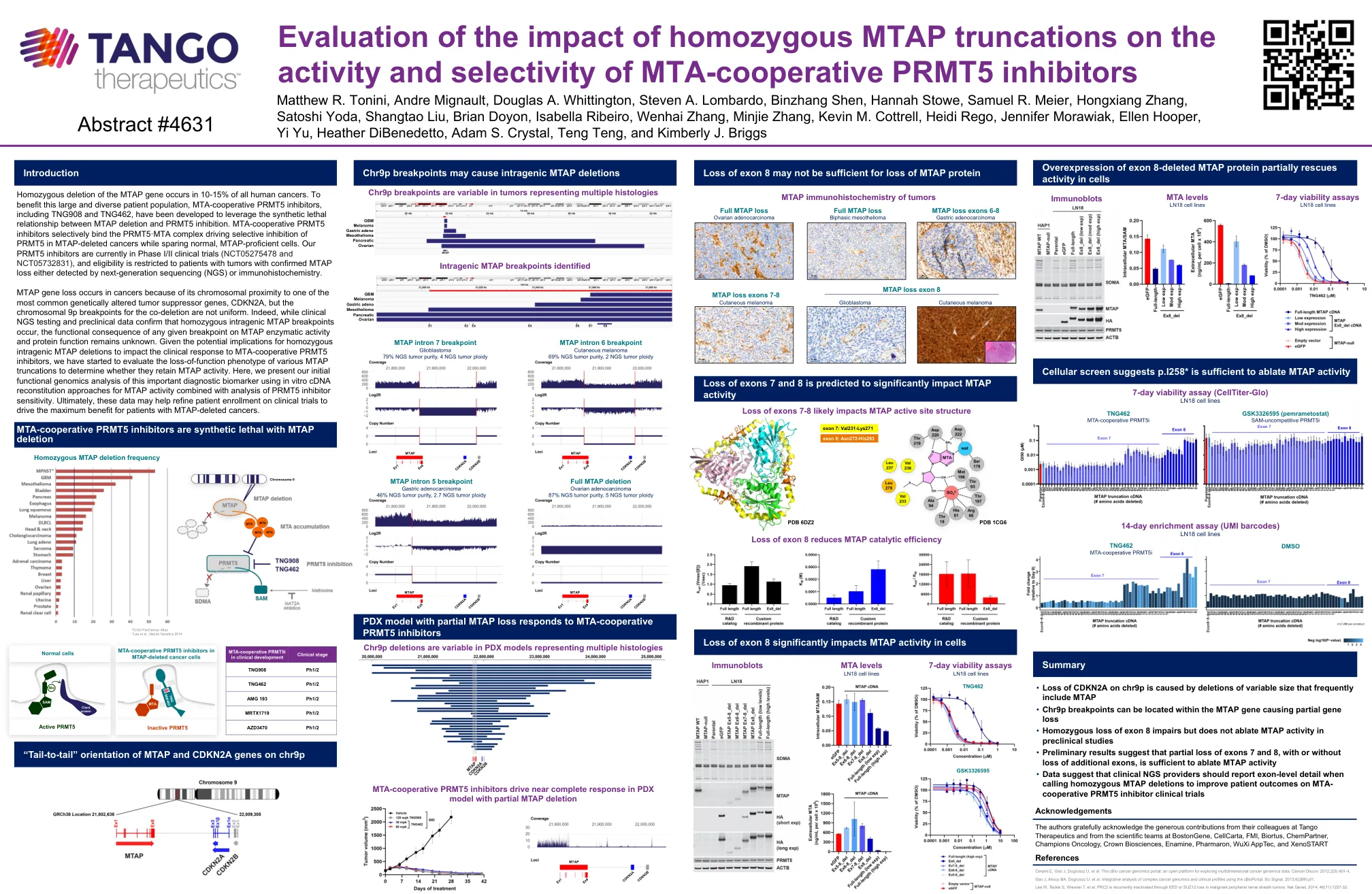
MTAP基因的纯合缺失发生在所有人类癌症中的10-15%中。为了使包括TNG908和TNG462在内的MTA合作PRMT5抑制剂受益,已开发出来的MTA合作PRMT5抑制剂,以利用MTAP缺失与PRMT5抑制之间的合成致死关系。MTA合并PRMT5抑制剂选择性地结合了PRMT5抑制剂在MTAP骨骼骨骼癌中的PRMT5驱动选择性抑制,同时保留正常的MTAP且可培养的细胞。我们的PRMT5抑制剂目前正在I/II期临床试验(NCT05275478和NCT05732831)中,并且资格仅限于具有确认的MTAP损失的肿瘤患者,即通过下一代测序(NGS)或免疫组织化学化学。mTAP基因丧失发生在癌症中,因为它与最常见的遗传改变的肿瘤抑制基因CDKN2A的染色体接近,但是共脱落的染色体9p断点并不均匀。的确,尽管临床NGS测试和临床前数据证实了纯合性内部MTAP断点,但MTAP酶活性和蛋白质功能上任何给定断点的功能后果仍然未知。鉴于对纯合基因内MTAP缺失的潜在影响会影响对MTA合件PRMT5抑制剂的临床反应,我们已经开始评估各种MTAP截断的功能丧失表型,以确定它们是否保留MTAP活性。在这里,我们使用体外cDNA重构方法与PRMT5抑制剂敏感性分析,对这种重要的诊断生物标志物进行了最初的功能基因组学分析。最终,这些数据可能有助于完善患者参加临床试验的入学率,以促进MTAP骨骼癌症患者的最大收益。
全基因组药物锚筛选将CAAP1和AKAP17A识别为PRMT5抑制剂敏感性的调节剂
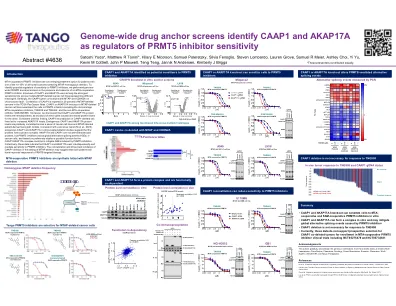
MTA合件PRMT5抑制剂是一种新兴治疗选择。确定对PRMT5抑制剂敏感性的潜在调节剂,我们在存在和不存在MTA合并PRMT5抑制剂的情况下进行了基因组宽CRISPR敲除筛选。敲除CAAP1和AKAP17A的敲除是代表不同组织学的多个MTAP缺失的癌细胞系中最强的致敏效果之一。引人注目的是,CAAP1基因在9p21染色体上与MTAP和CDKN2A共定位。在TCGA Pancancer Atlas中,有20%的MTAP删除癌症中有20%的CAAP1的删除CAAP1。CAAP1或AKAP17A在MTAP缺失的癌细胞系中的敲除将细胞敏感到PRMT5抑制剂,包括临床阶段的MTA合并抑制剂TNG908和TNG462,以及非MTA合作抑制剂GSK33226595。此外,我们发现CAAP1和AKAP17A蛋白水平是相互依存的,因为两个基因的敲除导致另一个基因的蛋白质水平降低。与这一发现一致,CAAP1在CAAP1删除细胞系中的重建导致AKAP17A水平升高。内源性CAAP1和AKAP17A蛋白水平在一系列癌细胞系和MTAP缺失的患者衍生异种移植模型之间呈正相关。与先前的报告(Ni等,2023),外源CAAP1和AKAP17A共免疫沉淀研究一致,这表明蛋白质形成蛋白质复合物。共同表明CAAP1和AKAP17A相互依存并介导对PRMT5抑制剂的敏感性。AKAP17A和CAAP1不是特征良好的蛋白质,但是PRMT5抑制剂在癌细胞中诱导全球替代剪接事件(ASE),基于初步研究,CAAP1/AKAP17A复合物的可能功能可以减轻PRMT5抑制引起的ASES。在MTAP缺失的情况下,共定位和CAAP1缺失的发生率为20%,可能表明此类患者对PRMT5靶向治疗的反应会提高。
Adam Yaari 公正的体外和体内药物锚筛网鉴定了MTAP-DEL中MTA合件PRMT5抑制剂的抗性和敏化机制 stearoyl-COA去饱和酶是SMAD4缺陷癌症中的合成致命靶标 发现TNG908:选择性,大脑渗透物,MTA MTA合件PRMT5抑制剂在MTAP中有效...
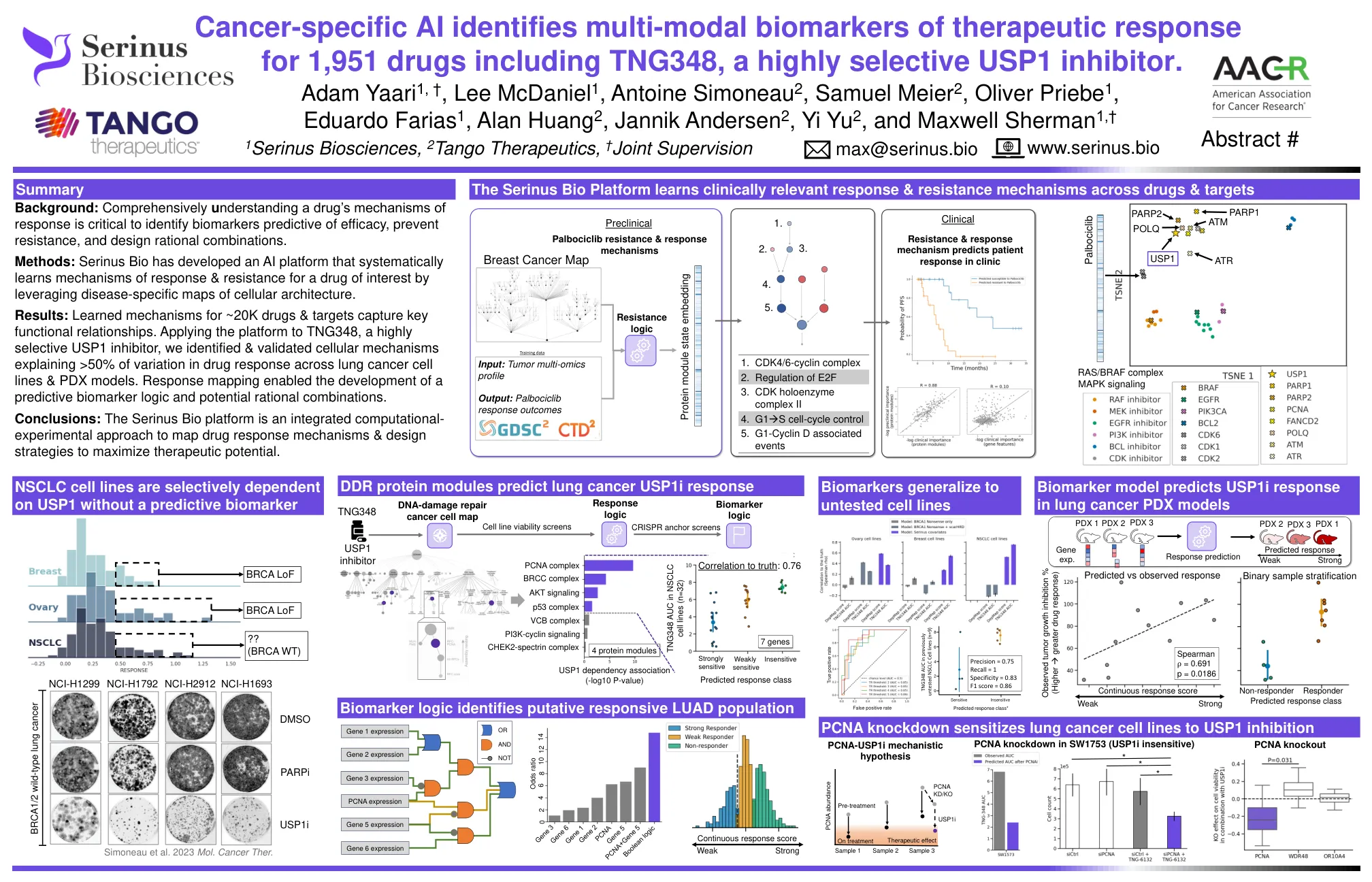
结果:〜20K药物和目标的学习机制捕获了关键的功能关系。将平台应用于高度选择性的USP1抑制剂TNG348,我们确定了和验证的细胞机制,解释了整个肺癌细胞系和PDX模型的药物反应变化> 50%。响应映射可以开发预测性生物标志物逻辑和潜在合理组合。