XiaoMi-AI文件搜索系统
World File Search SystemTyr


补充酪氨酸在压力或认知要求下对临床和健康人群的影响 - 审查
食用氨基酸酪氨酸(Tyr),多巴胺(DA)和去甲肾上腺素(NE)的前体,可能会抵消神经递质功能和认知性能的降低。然而,关于Tyr补充的有效性的报道差异很大,有些研究发现了有益的效果,而其他研究则没有。在这里,我们回顾了有关Tyr的可用认知/行为研究,以阐明补充Tyr是否可以对性能有益。使用Tyr补充来治疗临床疾病的潜力似乎有限,其受益可能会因神经递质功能受损和合成的存在和程度而确定。同样,补充剂增强体育锻炼的潜力似乎也很少,也许是因为体育锻炼与儿茶酚胺功能之间的联系是由许多其他因素介导的。相比之下,Tyr似乎确实有效地提高了认知表现,尤其是在短期的压力和/或认知要求的情况下。我们得出结论,Tyr是认知的有效增强子,但只有当神经递质功能完好无损并且DA和/或NE暂时耗尽时。©2015 Elsevier Ltd.保留所有权利。

氧化还原生物学
饮食原腺苷(PAC)消费量与结直肠癌(CRC)的风险降低有关。在包括CRC在内的人类癌症中,表皮生长因子(EGF)受体(EGFR)信号通路的失调频率很高。我们先前表明六聚体PAC(十六进制)在人CRC细胞中发挥抗增殖和凋亡作用。这项工作是否可以通过调节EGFR途径的能力发挥抗CRC效应。在增殖的CACO-2细胞中,十六进制的作用抑制了EGF诱导的EGFR二聚化和NADPH氧化酶依赖性磷酸化,在Tyr 1068时磷酸化,降低了EGFR在脂质筏上的位置,并抑制了促促进性和抗磷酸和抗蛋白质路线的下部激活RAF/MEK/ERK1/2和PI3K/AKT。在不存在和存在EGF的情况下, HEX还促进了EGFR的内在化。 虽然HEX在Tyr 1068时降低了EGFR磷酸化,但EGFR Tyr 1045磷酸化增加了。 后者为泛素连接酶C-CBL提供了一个对接位点,并通过溶酶体促进EGFR降解。 重要的是,HEX与靶向EGFR的化学治疗药物Erlotinib协同作用,均以降低EGFR磷酸化和抑制细胞生长的能力。 因此,饮食PAC可以通过通过氧化还原和非雷多斯调节的机制调节EGFR促进性信号通路来发挥抗CRC作用。 此外,十六进制还可以增强以EGFR为目标的药物的作用。HEX还促进了EGFR的内在化。虽然HEX在Tyr 1068时降低了EGFR磷酸化,但EGFR Tyr 1045磷酸化增加了。后者为泛素连接酶C-CBL提供了一个对接位点,并通过溶酶体促进EGFR降解。重要的是,HEX与靶向EGFR的化学治疗药物Erlotinib协同作用,均以降低EGFR磷酸化和抑制细胞生长的能力。因此,饮食PAC可以通过通过氧化还原和非雷多斯调节的机制调节EGFR促进性信号通路来发挥抗CRC作用。此外,十六进制还可以增强以EGFR为目标的药物的作用。
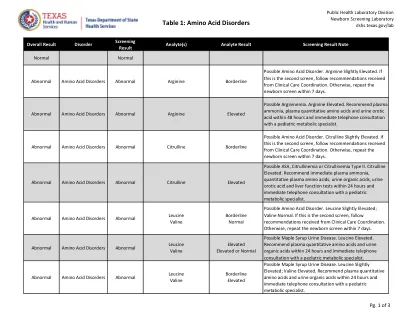
表1:氨基酸疾病 推广者(A)或社区卫生工作者(CHW)培训和... 如何审查免疫记录和德克萨斯州学校疫苗要求 表8:血红蛋白病 TVFC/ASN重新注册 负责您的健康 Texasaim Osud教师BIOS 记录疾病的历史:水痘(水痘) 新的novavax covid-19疫苗可用于订购 TVFC/ASN疫苗损失 - 报告浪费或过期... 疫苗分配和订购系统(VAO) 您可能对狂犬病不了解的8件事,但应 表1:德克萨斯州心血管疾病和中风会员的出席会议于2024年5月6日星期一举行。 unidos contra la糖尿病 负责您的健康 hep b妈妈 咨询委员会名称
可能的PKU,良性的热丙氨酸血症,辅因子生物合成或辅因子再生中生物蛋白缺损中的生物蛋白缺损。苯丙氨酸略有升高; phe/tyr抬高。在24小时内建议血浆定量氨基酸,并立即与儿科代谢专家进行电话咨询。

基于喹唑啉的 VEGFR-2 抑制剂作为潜在的抗...
ROS 活性氧 RPTEC 肾近端小管上皮细胞 SAR 构效关系 Sck 血清肌酸激酶 Src 肉瘤 TGI 肿瘤生长抑制 Thr 苏氨酸 Tie-2 血管生成素-1 受体 TSP 血小板反应蛋白 Tyr 酪氨酸 Val 缬氨酸 VEGF 血管内皮生长因子 VEGF-A 血管内皮生长因子 A VEGF-B 血管内皮生长因子 B VEGF-C 血管内皮生长因子 C VEGF-D 血管内皮生长因子 D VEGF-E 血管内皮生长因子 E VEGF-F 血管内皮生长因子 F


南卡罗来纳州 - 州健康评估代谢障碍
苯基酮(PKU)高苯基血症是一种氨基酸疾病,是由于活性降低,合成障碍或苯丙氨酸羟化酶或其辅助因子的降低或回收的氨基酸疾病,BH 4。苯基酮(PKU)是由苯丙氨酸羟化酶缺乏引起的。没有这种酶,身体无法将苯丙氨酸(PHE)转化为酪氨酸(Tyr)。 苯丙氨酸在血液,尿液和中枢神经系统中积聚。 如果未经治疗,婴儿将经历深刻的智力残疾(ID)。 她或他也可以减少皮肤和头发的色素沉着,发霉的气味,异常行为和/或癫痫发作。 筛查PKU还可以鉴定出患有良性多苯基丙氨酸血症的婴儿,生物蛋白质辅因子生物合成的缺陷和生物蛋白辅助剂再生的缺陷。 Inheritance: Autosomal recessive Estimated Incidence: PKU - 1:16,000 Benign hyperphenylalaninemia (H-PHE) - unknown Defects of biopterin cofactors biosynthesis (BIOPT-BS) or regeneration (BIOPT-REG) - unknown, thought to be very rare Abnormal Screen Result: Elevated PHE Elevated PHE/TYR Method of Notification: All critical results are打电话给记录提供商。 下一步如果异常:在滤纸上尽快重复新生儿屏幕。 在已知重复结果之前,没有公式/喂养更改。 如果PHE在重复标本中仍明显升高,请参阅儿科代谢专家。 可能需要进一步的诊断评估来排除BH 4缺陷。 代谢专家将与代谢营养师协调进行PHE限制饮食。没有这种酶,身体无法将苯丙氨酸(PHE)转化为酪氨酸(Tyr)。苯丙氨酸在血液,尿液和中枢神经系统中积聚。如果未经治疗,婴儿将经历深刻的智力残疾(ID)。她或他也可以减少皮肤和头发的色素沉着,发霉的气味,异常行为和/或癫痫发作。筛查PKU还可以鉴定出患有良性多苯基丙氨酸血症的婴儿,生物蛋白质辅因子生物合成的缺陷和生物蛋白辅助剂再生的缺陷。Inheritance: Autosomal recessive Estimated Incidence: PKU - 1:16,000 Benign hyperphenylalaninemia (H-PHE) - unknown Defects of biopterin cofactors biosynthesis (BIOPT-BS) or regeneration (BIOPT-REG) - unknown, thought to be very rare Abnormal Screen Result: Elevated PHE Elevated PHE/TYR Method of Notification: All critical results are打电话给记录提供商。下一步如果异常:在滤纸上尽快重复新生儿屏幕。在已知重复结果之前,没有公式/喂养更改。如果PHE在重复标本中仍明显升高,请参阅儿科代谢专家。可能需要进一步的诊断评估来排除BH 4缺陷。代谢专家将与代谢营养师协调进行PHE限制饮食。向SC新生儿筛查计划报告所有发现。新生儿介绍:无。然而,年龄,低出生体重和/或TPN可能会影响多种氨基酸水平,包括精氨酸,蛋氨酸,苯丙氨酸,缬氨酸和亮氨酸。治疗:生物蛋白质辅因子生物合成或再生的PKU/缺陷:PHE限制饮食的生命。特殊的代谢配方可供所有SC居民(并确认诊断),目前无需任何费用。BH 4缺陷需要其他诊断评估和治疗。

CRISPR/Cas9 介导双等位基因 F0 海葵鱼 (Amphiprion ocellaris) 突变体的产生
基因组操作是一种有用的方法,可用于阐明发育、生理和行为方面的分子途径。然而,由于缺乏适用于珊瑚鱼的基因编辑工具,因此它们许多独特特征的遗传基础仍有待研究。一种适合应用这种技术的标志性珊瑚鱼群是海葵鱼 (Amphiprioninae),因为它们与海葵共生、雌雄同体、复杂的社会等级、皮肤图案发展和视觉,并且相对容易在水族箱中饲养,因此被广泛研究。在这项研究中,我们开发了一种基因编辑方案,用于将 CRISPR/Cas9 系统应用于眼斑海葵鱼 (Amphiprion ocellaris)。受精卵的显微注射用于证明我们的 CRISPR/Cas9 方法在两个不同靶位点的成功应用:与视觉有关的视紫红质样 2B 视蛋白编码基因 (RH2B) 和与黑色素生成的酪氨酸酶生成基因 (tyr)。对眼斑海马胚胎中测序的靶基因区域进行分析表明,注射胚胎的吸收率高达 73.3%。进一步分析亚克隆的突变基因序列并结合扩增子散弹枪测序表明,我们的方法在 F0 眼斑海马胚胎中产生双等位基因突变的效率为 75% 到 100%。此外,我们清楚地显示了 tyr 突变胚胎的功能丧失,其表现出典型的低黑色素表型。该方案旨在作为进一步探索 CRISPR/Cas9 在眼斑海马中潜在应用的有用起点。眼斑鱼,作为研究小丑鱼和其他珊瑚鱼基因功能的平台。

优化基因治疗的可扩展RAAV生产
• Data from a perfusion benchtop bioreactor scale show that Ala, Asn, His, Tyr, and Trp positively correlate with capsid production, while Gln correlates negatively • Analysis of cell culture media from shake flask experiments shows that Asn and other amino acids, including several essential amino acids, deplete during AAV production • Capsid titer per cell increases when Gln is absent from media • The REBEL device enabled通过准确,精确地定量氨基酸的数据驱动的RAAV生产的生物过程开发,用于HEK293细胞中的RAAV生产,从而进一步了解过程•正在进行更多的实验,以优化灌注过程,以获得全面的Capsids

评估 CRISPR-Cas9 在生成中的效率......
a.bonillap@uniandes.edu.co 摘要 - 神经连接蛋白 (NL) 是跨突触细胞粘附蛋白,当它们与突触前神经连接蛋白相互作用时,参与形成突触。越来越多的证据表明它们与某些神经精神疾病有关,这使它们成为研究的相关蛋白质。神经连接蛋白-1 基因 (nlgn1) 是 NL 家族的一员,参与兴奋性突触,与压力相关疾病(重度抑郁症和创伤后应激障碍)的遗传基础有关。然而,它在压力调节中的具体作用尚未得到详细研究。斑马鱼 (Danio rerio) 已成为转化神经科学和行为研究的模型,可用于研究与焦虑、恐惧、睡眠、压力和记忆相关的行为,使其成为此类研究的理想模型。在这里,我们旨在使用 CRISPR-Cas9 系统生成 nlgn1 缺陷斑马鱼,并评估这种基因改造对幼虫行为的影响。为此,我们必须标准化 CRISPR 基因编辑系统。我们最初使用 CRISPR-cas9 生成 tyr -crispant 斑马鱼胚胎,因为这会产生可见的表型。我们用 tyr 验证了我们的 CRISPR 基因编辑系统,产生了具有不同程度嵌合性的 crispant 样表型,效率约为 76%。然后,注射了针对 nlgn1 基因的 sgRNA 的斑马鱼胚胎的存活率较低,在 11.9% 到 7% 之间,具体取决于所使用的 sgRNA。此外,许多胚胎在发育过程中出现畸形,并在 3dpf 时致命。在旷场测试期间,nlgn1 -crispant 和对照组之间的不动性平均值有显著差异。这些结果表明,神经连接蛋白-1 可能在发育过程中发挥关键作用或具有强烈的神经表型。