XiaoMi-AI文件搜索系统
World File Search System剥皮

在大型纯种牲畜种群中使用杂交剥皮进行全基因组序列推断的准确性
摘要背景:合适的测序策略与填补方法的结合对于从牲畜种群中收集用于研究和育种的大型全基因组序列数据集至关重要。在本文中,我们描述并验证了测序策略与填补方法混合剥离在真实动物育种环境中的结合。方法:我们使用了四个不同规模的猪种群的数据(18,349 到 107,815 头猪),这些猪种群的基因分型广泛,全基因组标记密度在 15,000 到 75,000 个之间。每个种群中大约有 2% 的个体进行了测序(大多数为 1 × 或 2 ×,每个种群有 37–92 个个体,总计 284 个,为 15–30 × )。我们使用混合剥离技术填补了全基因组序列数据。我们使用留一法设计,通过删除覆盖率高的 284 个个体的序列数据来评估填补准确性。我们模拟了模仿真实人群中使用的测序策略的数据,以使用回归树量化影响个体和变异插补准确性的因素。结果:四个人群中大多数个体的插补准确性都很高(个体剂量相关性中位数:0.97)。由于缺乏自身和祖先的标记阵列数据,每个人群最早几代个体的插补准确性低于其他人群。决定个体插补准确性的主要因素是基因分型状态、直系祖先的标记阵列数据的可用性以及与其他人群的关联程度,但亲属的测序覆盖率没有影响。决定变异插补准确性的主要因素是次要等位基因频率和每个变异位点具有测序覆盖的个体数量。通过实证观察验证了结果。结论:我们证明,将适当的测序策略与混合剥离相结合是一种强大的策略,可以在大型谱系群体中生成高精度的全基因组序列数据,其中只有一小部分个体(2%)进行了测序,而且大部分覆盖率较低。这是成功实施全基因组序列数据进行基因组预测和精细定位因果变异的关键步骤。

七lamp虫的正常发育阶段,Lampetra重新发行(dybowski)
在本世纪中叶,大湖渔业遭受了灾难的威胁,起源于寄生lamp鼠,彼得罗森·马里纳斯(Petromyzon Marinus)的数量。旨在控制这种威胁的研究计划中,Piavis负责检查温度对该物种中胚胎发育的影响[11]。他还进行了材料胚胎发育的阶段,因为标准阶段的确定对于更好地理解实验结果至关重要[11,12]。piavis细分了从排卵但未剥皮的卵到ammocoete幼虫的第一阶段到19个阶段的发展过程。

您的生物多样性指南汤森自然公园
Rambutan是属于Sapindaceae家族的本地热带果树。其他热带果树(如lychee,Longan和Pulasan)属于同一家族,并且与Rambutan密切相关。Rambutan的果实在其鲜红色的外观上具有毛茸茸的突起。剥皮时,揭示了一个白色多汁的肉。rambutan树是常绿的,成熟的树木每年最多可产生90公斤水果。濒临灭绝的莱佛士带来的兰格(Presbytis股骨)和罕见的长尾长尾小鹦鹉(Psittacula Longicauda)以果实为食。



虚拟现实作为可能的老年护理技术
虚拟现实(VR)是一种可以将其定义为“三维计算机生成的模拟环境,它试图复制现实世界或想象中的环境和互动,从而支持工作,教育,娱乐和健康”(Abbas等,2023,第7页)。在以医疗保健为中心的系统评价(Abbas等,2023)中出现了这种相对广泛的定义,这是本研究文章的领域,但在整个文献中都使用了其他定义(例如,请参见Burdea和Coiffet,2003; Furht,2008; Heim,2008; Heim,2008; Heim,1993; Heim; Heim; Heim; 1993; 1993; Steuuer,2000年)。vr可以是非剥皮或沉浸式的(Hamad and Jia,2022; Wohlgenannt等,2020),前者以围绕用户的筛选形式(Rahouti等,2021),而后者通过使用(HMDS)(HMDS)的使用,而后者是
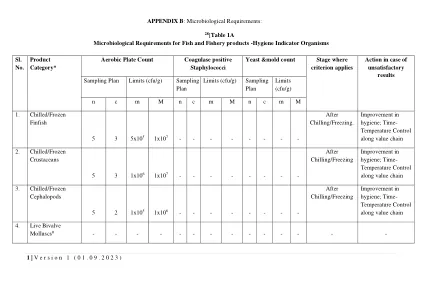
版本1(01.09.2023)附录B:微生物学...
良好的制造实践。寒冷是将鱼类或鱼类制品冷却到融化冰的温度的过程。可以通过使用冰,冷藏水,海水和淡水或冷藏海水的冰浆来实现寒冷。同样,冻结是足以将整个产品温度降低到足够低的水平,以保持鱼的固有质量,并且在运输,存储和分配期间一直保持在低温,直至最终销售的时间。以适当的设备进行的冻结过程,以使最大结晶的温度范围迅速通过。除非和直到产物温度达到–18 o C(0 O F)或在热稳定后的热中心,否则不得将快速冷冻过程视为完整。(2)冰冷/冷冻的甲壳类动物包括清洁,整体或剥皮的甲壳类动物(虾/虾,螃蟹和龙虾),它们在生,冷冻或冷冻
案例研究 - 更好的建筑物解决方案中心
每小时100,000磅(PPH)生物量锅炉为两个贴面干衣机,23个对数条件隧道和发电发电生成蒸汽。蒸汽还用于产生热水以在剥皮之前剥落原木。干燥单板所需的300 psig蒸汽也源自锅炉。干燥机的闪光蒸汽用于增加蒸汽供应到单板干燥过程。单板干燥机排气被捕获,并将其重新安装到锅炉上,以用作燃烧空气。贴面干燥器通常使用总蒸汽容量的30-40%,并且将剩余的蒸汽重新装饰到涡轮耦合发电机以产生电力。该植物每天消耗360吨木本生物量(每年131,400吨),其中一半来自两个不同的铣削操作的地点。剩余的生物质是在外部购买的。

设施简介
背景 ARBEC FOREST PRODUCTS INC. 产品 FORESTIERS ARBEC INC. (ARBEC) 购买了位于米拉米奇市的定向刨花板 (OSB) 工厂,该工厂原由 Weyerhaeuser Company Limited 拥有和经营。OSB 工厂于 1996 年投入使用,并以 Eagle Forest Products 的名义开始运营。Weyerhaeuser 随后于 2000 年 6 月购买了该工厂,并运营该设施直到 2007 年 1 月工厂因市场状况而关闭。ARBEC 于 2012 年秋季开始运营 OSB 工厂。米拉米奇的工厂生产尺寸为 4 英尺 x 8 英尺的 OSB 板。OSB 板主要用于住宅建筑。面板用于墙面护套、屋顶和结构地板。米拉米奇生产的大部分产品销往加拿大和美国。该工厂约有 150 名员工。工艺描述 简介 在米拉米奇的 OSB 工厂,所有木材都以圆木的形式通过卡车运送到现场,通常长度为 8 英尺。圆木通过两个自清洁闭环热池之一进入工厂,开始制造过程。热池的作用是在剥皮前松开木材上的树皮,并在冬季解冻冻结的原木。从热池出来的木材进入两个环形剥皮机之一,以去除原木上的树皮。然后,在三个刨片机之一中,将原木切成大约 0.03 英寸厚的小木条。木条在三个单程干燥机之一中干燥,其中刨片的含水量从 75 - 100 % 降低到 1.5 - 3 %。干燥的刨片进入两个大直径滚筒混合机之一,在那里与乳化蜡和液态树脂混合。然后,薄片在成型机上被排列成层,然后在高压和高温下压制以形成定向刨花板。然后将板切割成合适的尺寸,包装和储存,然后运送给客户。压机、热池和一般建筑物的热量是由炉中木材残余物的燃烧产生的。下面提供了热能系统、干燥机和空气污染控制设备的更详细描述。热能系统剥皮过程中产生的所有树皮和湿木材残余物都作为燃料在燃木炉中燃烧,为工厂产生热量。燃木炉由 GTS Energy Systems 制造,热额定值为 8650 万 kJ/小时(8200 万 BTU/小时)。轻油(#2 燃料油)用作 GTS 炉的备用燃料。燃木炉燃烧室内的温度保持在 450°C 至 1000°C(842°F 至 1832°F)之间。来自燃烧室的热气体通过一个系统来加热加热线圈内的导热油。加热后的导热油被泵送到各种