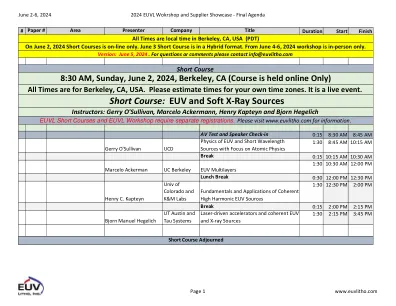
XiaoMi-AI文件搜索系统
World File Search System化学计算

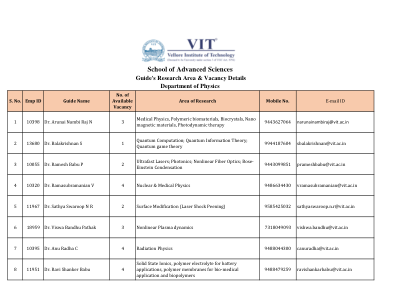
使用基于片段的量子化学计算蛋白质-配体相互作用能量的收敛协议
基于碎片的量子化学方法提供了一种避免电子结构计算的非线性缩放的方法,因此可以使用高级方法研究大型分子系统。在这里,我们使用碎片来计算具有数千个原子的系统中的蛋白质-配体相互作用能,使用一种用于管理基于碎片的计算的新软件平台,该平台实现了屏蔽多体展开。使用最小基半经验方法 (HF-3c) 进行的收敛测试表明,使用单残基碎片和简单氢帽的二体计算足以重现使用传统超分子电子结构计算获得的相互作用能,误差在 1 kcal/mol 以内,计算成本约为 1%。我们还表明,HF-3c 结果说明了密度泛函理论在增强四倍 ζ 质量的基组中获得的趋势。碎片化的战略部署有利于融合生物分子模型系统与高质量电子结构方法和基组一起使用,将从头算量子化学引入迄今为止难以想象的规模的系统。这将有助于为机器学习应用生成高质量的训练数据。


结合机器学习和量子化学计算进行热激活延迟荧光分子材料的高通量虚拟筛选:选择策略和结构突变的影响
以重过渡贵金属有机配合物(如Ir(III)的联吡啶配合物)为代表的磷光材料,直到第三代TADF材料(如有机给体-p桥-受体分子)。在电激发下,TADF材料(以非常低的第一激发单重态-三重态能隙(DE ST)为特征的化合物)被热激活,以诱导有效的逆系间窜越(rISC),其中三重态激子转化为单重态激子,从而主要从发射的单重态激发态发光。图1示意性地示出了TADF材料的电致发光过程。与贵金属有机配合物磷光材料相比,TADF材料具有材料空间更大、价格低廉、易于制备和合成、易于制作柔性屏幕以及蓝光发射更稳定的优势。因此,近十年来,作为现代OLED最有前途的电致发光材料,它们得到了实验2,5 - 9 、理论10 - 23 和理论-实验相结合15,24,25的深入研究。基本上,有两类TADF材料得到了认真探索4。第一类是纯有机D - A或D - p - A体系,其电子给体(D)或受体(A)主要由含氮芳香杂环构成。最低激发态通常具有显著的分子内电荷转移(CT)跃迁特性。经过合理的设计和优化,基于此类TADF材料的OLED器件的外量子效率(EQE)甚至可以高达30%。从结构特征上看,由于给体和受体部分之间有足够的空间位阻,最好的发光效率通常对应于扭曲的D – A(或D – p – A)化合物。另一类是电子排布为d 10 的过渡金属(Cu(I)、Ag(I)、Zn(II)等)配合物,它们的最低激发态通常具有明显的金属 – 配体电荷转移(MLCT)跃迁特征。饱和的d 10

设计光敏化合物的新型量子经典计算方案可以加速材料发现
最近,量子化学计算与机器学习的结合在加速新材料发现方面表现出了巨大的潜力。虽然这种混合方法与传统方法相比消耗的资源和时间更少,但它仍然面临着根本性的挑战。这些挑战包括训练数据集的大小和质量限制,以及使用离散优化技术有效探索大型化学空间的困难。

地球科学家的微生物学
本书的偏见是基于我自己的经验和兴趣。我在整本书中使用了许多示例。这个选择不是因为我认为我的研究优于其他例子,而是因为它通常是我的方便选择。在整本书中,我还强调地球化学。在这方面,微生物在很大程度上是基于其功能(例如硫酸盐还原器)而不是其分类学描述的。此外,该书详细描述了一些地球化学计算,但仅给出了一个非常简短的示例,说明了如何使用分子技术分析微生物群落。这个选择反映了我作为研究人员的优势,但它也是地球科学家的微生物学方面的方便途径,地球科学家通常熟悉地球化学但不是分子生物学。


