XiaoMi-AI文件搜索系统
World File Search System合件



公告第124号 合和6年10月15日
2024年10月9日 — (3)投标项目的规格应为项目明细表规格栏中指定的规格,或相当或更优(包括其他公司的产品)。 (4)如果您正在竞标类似物品,请...

公告第8号 合和6年2月27日
2024 年 3 月 5 日 — 规格编号。4QGB10202070。4RL81CA0001 0001。产品名称或主题。Senso (6) 业务用一般废弃物处理(可燃物)。零件编号或规格。根据规格。要使用的设备名称。计划数量。

发现TNG908:选择性,大脑渗透物,MTAMTA合件PRMT5抑制剂在MTAP中有效...MTA合件PRMT5抑制剂在MTAP中有效...
目标:恶性外周神经鞘肿瘤(MPNST)是高度侵略性的麦芽瘤,治疗率有限,存活率较差,因此需要发展新的治疗疗法。由于由近端抑制基因CDKN2A损失造成的乘客缺失,大约25-50%的MPNST港口丧失酶甲基腺苷磷酸化酶(MTAP)损失。PRMT5由于底物甲基噻吩并腺苷(MTA)的积累而被鉴定为MTAP被骨化细胞中的选择性依赖性,该细胞本身就是内源性PRMT5抑制剂。TNG908和TNG462是临床阶段MTA合件PRMT5抑制剂,可分别证明对MTAP细胞的选择性分别在15倍和45倍的MTAP -INTACT -INTACT细胞上。先前的报道表明,这两个分子都驱动了各种MTAP癌症组织学的异种移植模型中耐用的肿瘤回归。在这里,我们的目标是检查临床前MPNST模型中TNG908和TNG462的活性。

基础临床社会医学统合讲义
09:00-09:50 Tadaki(国家传染病研究所)感染性病理学对Covid-19的贡献10:00-10:00-10:50 Yamazaki Akira(大阪大学)(大阪大学)细胞介导的免疫反应对SARS-COV2 11:00-11:00-11:00-11:00-11:50 ARASE NAO(OSAKA NAO)介绍了OSAKA NAO(OSAKA NAO),以下简13:00-13:50 Nishiura Hiroshi(京都大学)Covid -19的传染病流行病学194:00-14:50 Sato Yoshi(Tokyo)新颖的Coronavirus大学的演变15:00-15:00-15:50-15:50

冰箱和 1 件其他物品
7. 注意事项 (1) 参赛资格相关事项 A. 应征者不得存在《主计法》第七十条规定的情况。此外,未成年人、被监护人或接受协助的人,即使已经取得订立合同所必需的同意,也属于同一条款内有特殊事由的情况。 (一)不属于预算会计审计法第七十一条规定情形的。 参加国防部招标的资格(各部委统一资格)为2022、2023、2024财年。 (e)目前不受日本内阁官房长官、防卫政策局局长、采购技术后勤局局长或陆上自卫队参谋长根据“设备等及服务采购暂停提名指南”暂停提名的限制。 e. 与前项规定暂停指定之人有资本或个人关系,且无意与国防部签订与该人相同种类之物品买卖、制造或承包服务契约之人。 原则上,已被暂停投标资格的人员不得进行分包。但有关部会暂停提名权机关认定确有不可避免的情况时,不在此限。 (k)投标人审查完招标和合同指南后,应在投标书中附加一份声明:“我公司特此承诺遵守招标和合同指南中关于排除有组织犯罪的项目。”


1 2 3 序号 主题 交付(履行) 地点 交付日期(...
判定方法为该金额的10%(减税对象品为8%,若该金额有不足1日元的尾数,则四舍五入),并输入估价的110/100(减税对象品为108/100)。以团队设定的估价范围内的最低估价为判定标准。如果有两名或两名以上的申请人提供最低出价,则以抽签方式确定申请人。 6. 创建合同
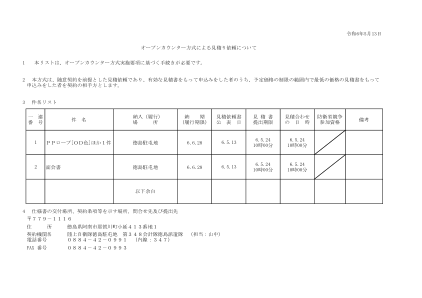
1 2 3 一连番号件名纳入(履行) 场所纳期( ...
估算金额将根据上述内容确定(减税对象品为 8%,金额小数部分不足 1 日元时,小数部分四舍五入)。因此,无论估算者是消费税、地方消费税的纳税人还是免税人,估算者都必须在估算中输入估算金额的 110/100(减税对象品为 108/100)。估算者将在本机构规定的估算价格范围内提供最低估算金额。如果有两名或两名以上的申请人提供最低出价,则将通过抽签方式确定申请人。 6. 准备合同 合同金额50万日元以上时,准备发票,150万日元以上时,准备合同。