XiaoMi-AI文件搜索系统
World File Search System扩增子

23-023-02cr:请求
利用选择标记鉴定转化植物,并筛选 T-DNA 拷贝数。通过扩增子测序鉴定编辑的 T0 植物(Clement 等人,2019 年;Illumina MiSeq 系统指南,2018 年;Illumina MiSeq 系统指南,2019 年),自交,并通过扩增子测序分析所得的 T1 植物以确认编辑。进行了额外的 PCR 检测,以确认不存在 T-DNA 插入物和质粒骨架(Applied Biosystems 用户公告 #2)。选择包含所需编辑但没有 T-DNA 或质粒骨架的纯合 T1 植物 P227933.30 进行延续,并将其命名为 GM200007。T1 植物不含外来 DNA,在 [ ] 基因中含有纯合缺失。表 1. 用于创建 GM200007 大豆的转化载体 F137620 的遗传元件


使用基于标记扩增子的 NGS 检测方法通过液体活检基因组分析确定具有可操作突变的致癌成瘾晚期 NSCLC 患者的预后
基于循环肿瘤 DNA (ctDNA) 的分子分析正在通过多基因下一代测序 (NGS) 面板在晚期癌症患者的临床实践中迅速获得关注。然而,临床结果仍然描述不详,需要通过对血浆 ctDNA 中检测到基因组改变的患者进行个性化治疗来进一步验证。在这里,我们描述了通过 ctDNA 液体活检检测 InVisionFirst ® -Lung 在血浆中发现可操作改变的致癌成瘾晚期 NSCLC 患者的结果、3 个月时的疾病控制率 (DCR) 和无进展生存期 (PFS)。对 81 名晚期 NSCLC 患者进行了汇总回顾性分析,这些患者具有预测对目前 FDA 批准药物有反应的所有类型的改变:致敏常见 EGFR 突变(78%,n = 63)和 T790M(73%,46/63)、ALK / ROS1 基因融合(17%,n = 14)和 BRAF V600E 突变(5%,n = 4)。所有患者均通过先前的组织基因组分析确认了液体活检中检测到的可操作驱动改变,并且所有患者都接受了个性化治疗。在接受匹配靶向治疗的 82 名患者中,10% 为一线患者,41% 为二线患者,49% 为二线以上患者。 73% (46/63) 的患者在 TKI 复发时被检测到获得性 T790M,所有潜在患者 (34/46) 均根据 ctDNA 结果开始奥希替尼治疗。81 名可评估患者的 3 个月 DCR 为 86%。中位 PFS 为 14.8 个月 (12.1-22.9 个月)。基线 ctDNA 等位基因驱动基因分数与个性化治疗的反应率无关 (p = 0.29)。ctDNA 分子分析是一种准确可靠的工具,可用于检测晚期 NSCLC 患者中临床相关的分子改变。靶向治疗的临床结果支持将基于扩增子的 NGS ctDNA 分析液体活检用于晚期 NSCLC 患者的一线和复发检测。

使用基于标记扩增子的 NGS 检测方法通过液体活检基因组分析确定具有可操作突变的致癌成瘾晚期 NSCLC 患者的预后
基于循环肿瘤 DNA (ctDNA) 的分子分析正在通过多基因下一代测序 (NGS) 面板在晚期癌症患者的临床实践中迅速获得关注。然而,临床结果仍然描述不详,需要通过对血浆 ctDNA 中检测到基因组改变的患者进行个性化治疗来进一步验证。在这里,我们描述了通过 ctDNA 液体活检检测 InVisionFirst ® -Lung 在血浆中发现可操作改变的致癌成瘾晚期 NSCLC 患者的结果、3 个月时的疾病控制率 (DCR) 和无进展生存期 (PFS)。对 81 名晚期 NSCLC 患者进行了汇总回顾性分析,这些患者具有预测对目前 FDA 批准药物有反应的所有类型的改变:致敏常见 EGFR 突变(78%,n = 63)和 T790M(73%,46/63)、ALK / ROS1 基因融合(17%,n = 14)和 BRAF V600E 突变(5%,n = 4)。所有患者均通过先前的组织基因组分析确认了液体活检中检测到的可操作驱动改变,并且所有患者都接受了个性化治疗。在接受匹配靶向治疗的 82 名患者中,10% 为一线患者,41% 为二线患者,49% 为二线以上患者。 73% (46/63) 的患者在 TKI 复发时被检测到获得性 T790M,所有潜在患者 (34/46) 均根据 ctDNA 结果开始奥希替尼治疗。81 名可评估患者的 3 个月 DCR 为 86%。中位 PFS 为 14.8 个月 (12.1-22.9 个月)。基线 ctDNA 等位基因驱动基因分数与个性化治疗的反应率无关 (p = 0.29)。ctDNA 分子分析是一种准确可靠的工具,可用于检测晚期 NSCLC 患者中临床相关的分子改变。靶向治疗的临床结果支持将基于扩增子的 NGS ctDNA 分析液体活检用于晚期 NSCLC 患者的一线和复发检测。

Illumina数据分析指南
Illumina在Imlumina平台上测序在Illumina平台上测序的cleanplex amplicon库工作流程设置建议建议使用Illumina的本地运行经理(LRM)DNA Amplicon分析模块进行分析。LRM DNA扩增子分析模块v2.1.0可在ISEQ(控制软件V1或V2),Miseq(MCS V3),NextSeq 500/550(NCS 4.0)和NextSeq 500DX(NCS 4.0)上获得。LRM DNA扩增子分析模块v3.0.0.14可在Miseq(MCS v4.0)上获得。这些说明是一个简单的摘要,描绘了如何为已经完成的运行设置分析,或设置新运行以包括分析。样品必须通过基因组进行分析以进行分析:如果在一个测序运行中存在不同的基因组,则只能设置新运行以用一个基因组进行运行分析。但是,这些样品可以在同一运行中进行测序,然后通过基因组分批分析。如果测序运行中的所有样本均来自同一基因组,则用户可以使用任何一种分析方法。此模块对齐扩增子读取针对清单文件中指定的参考,并要求变体针对目标区域。工作流程还产生了运行质量和覆盖信息的摘要报告。有用的提示和资源:●有关其他详细信息和故障排除,请参阅Illumina的最新本地运行经理DNA

Trusight-Concology-500 and-Ht-Data-Sheet- ...
库制备基于经过验证的杂交捕获化学,可从基于DNA和RNA的库中纯化选定的靶标。生物素化的探针与感兴趣的区域杂交,这些区域使用链霉亲和素涂层的磁珠将其拉下,然后洗脱以丰富库池。基于杂交的富集是一种有用的策略,用于分析给定样品中的特定遗传变异,并可靠地测序外部或大量基因(例如,> 50个基因)。它在广泛的输入类型和数量上提供可靠的结果。混合捕获化学具有比扩增子测序的几个优点,包括产生较少的伪影和辍学的数据。此外,杂交捕获化学是融合不可知的,可以检测和表征已知和新型融合。与基于扩增子的方法不同,该方法需要确认性测试,因为可能会出现假阳性,而混合捕获方法高度敏感,并且可以准确地表征已知和新型伴侣的基因融合。

顶部4 -Bio -me
基于扩增子的NGS针对细菌16S rRNA基因或真菌ITS1区域,传统上用于阴道微生物组分析。尽管此方法具有成本效益,但它不适合可靠地解决物种水平,实际上通常仅限于属水平分辨率。最近在NGS平台上的突破是长阅读技术的开发,例如牛津纳米孔技术提供的技术。尽管这些平台启用了全长16S rRNA基因扩增子测序,但已证明在物种水平上提供了更好的分辨率,而不是短阅读技术4,而长阅读平台往往每个基础准确性,较高的总成本和较低的吞吐量5,6。相反,shot弹枪宏基因组测序目标不仅是16S rRNA基因/真菌ITS1区域,而且是微生物组的集体基因组信息,该信息允许在该物种和应变水平3,7上进行分类识别。
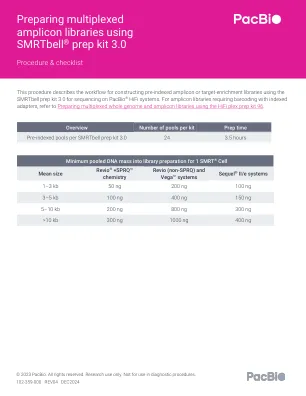
使用 SMRTbell® 准备多重扩增子文库...
使用凝胶提取的扩增子产物可能会导致测序性能降低,这是因为插入染料(如溴化乙锭)和暴露于紫外线辐射会造成固有的损坏。如果使用已用染料染色的凝胶提取产物,建议在文库制备和测序之前对其进行额外的扩增,以去除损坏和/或染料。

高效的敲入方法能够实现谱系追踪...
在这里,我们设计了一种无需克隆的 39 敲入策略,用于斑马鱼,使用 PCR 扩增的 dsDNA 供体,避免破坏目标基因。dsDNA 供体携带编码荧光蛋白和 Cre 重组酶的遗传盒,与内源基因同框,但被可自裂解的肽与其隔开。具有 59 AmC6 末端保护的引物产生了具有更高整合效率的 PCR 扩增子,这些扩增子与预组装的 Cas9/gRNA 核糖核蛋白复合物共注射以进行早期整合。我们针对四个基因位点(krt92、nkx6.1、krt4 和 id2a)并生成了 10 个敲入系,它们可作为内源基因表达的报告基因。敲入 iCre 或 CreERT2 的细胞系用于谱系追踪,结果表明 nkx6.1 + 细胞是多能性胰腺祖细胞,逐渐局限于双能性导管,而 id2a + 细胞在肝脏和胰腺中都是多能性细胞,逐渐局限于导管细胞。此外,肝脏 id2a +