XiaoMi-AI文件搜索系统
World File Search System这批

海报:resDNASEQTM HEK293 和 E1A 片段长度定量分析:基因治疗和疫苗制造中 GMP 批签发的综合解决方案
目的生物制药产品必须限制宿主细胞残留 DNA 污染物,以防止对患者产生基因毒性和免疫毒性风险。现有的残留宿主细胞 DNA 监管指南要求每剂量 ≤10ng,DNA 大小为 200bp 或更低。在病毒载体生产中,衣壳化 DNA 的数量、大小和致癌序列是额外的关注点。为了满足这一需求,赛默飞世尔科技开发了两种旨在满足监管指导的互补检测方法。Applied Biosystems™ resDNASEQ™ 定量 HEK293 DNA 试剂盒可定量总残留宿主细胞 DNA,Applied Biosystems™ resDNASEQ™ E1A DNA 片段长度试剂盒可对针对 E1A 致癌基因的短(86bp)、中(200bp)和长(476bp)片段进行大小分析。

和黄医疗宣布沃利替尼新药申请在中国获批,用于治疗初治或
香港、上海和新泽西州弗洛勒姆帕克 — 2024 年 3 月 28 日星期四:和记医疗(中国)有限公司(“和记医疗”)(纳斯达克/AIM:HCM;港交所:13)今天宣布,沃利替尼的补充新药申请(“sNDA”)已被中国国家药品监督管理局(NMPA)受理,用于治疗间充质上皮转化因子(“MET”)外显子 14 跳跃变异的局部晚期或转移性非小细胞肺癌(“NSCLC”)成人患者。如获批准,沃利替尼的新标签适应症将扩大到包括中国未接受过治疗的患者。此前,沃利替尼已在中国有条件批准用于治疗 MET 外显子 14 跳跃变异的 NSCLC 患者,这些患者在先前的全身治疗后病情进展或无法接受化疗。沃利替尼由我们的合作伙伴阿斯利康针对该患者群体推出并以商品名ORPATHYS®进行营销,这是首个在中国获批的选择性MET抑制剂。全球超过三分之一的肺癌患者在中国,而在全球NSCLC患者中,约有2-3%的肿瘤存在MET外显子14跳跃变异。验证性IIIb期临床试验(NCT04923945)一线队列的初步疗效和安全性数据将于2023年9月在IASLC世界肺癌大会(WCLC)上公布。验证性IIIb期试验的最终数据将于2024年3月20日在欧洲肺癌大会上公布。该研究数据为沃利替尼作为未经治疗或既往接受过治疗的MET外显子14跳跃变异NSCLC患者的靶向治疗选择提供了验证性证据。独立评审委员会评估显示,在未接受治疗的患者中,客观缓解率(“ORR”)为 62.1%(95% CI:51.0% 至 72.3%),疾病控制率(“DCR”)为 92.0%(95% CI:84.1% 至 96.7%),中位缓解持续时间(“DoR”)为 12.5 个月(95% CI:8.3 个月至 15.2 个月)。中位无进展生存期(“PFS”)为 13.7 个月(95% CI:8.5 个月至 16.6 个月),中位总生存期(“OS”)未达到,中位随访时间为 20.8 个月。在既往接受过治疗的患者中,独立审查委员会评估的 ORR 为 39.2%(95% CI:28.4% 至 50.9%),DCR 为 92.4%(95% CI:84.2% 至 97.2%),中位 DoR 为 11.1 个月(95% CI:6.6 个月至未达到)。中位 PFS 为 11.0 个月(95% CI:8.3 个月至 16.6 个月),中位 OS 尚未成熟,中位随访期为 12.5 个月。无论是初治患者还是既往接受过治疗的患者,反应都出现得很早(反应时间为 1.4-1.6 个月)。安全性是可以容忍的,没有观察到新的安全信号。最常见的 3 级或以上药物相关治疗出现的不良事件(占患者 5% 或更多)为肝功能异常(16.9%)、丙氨酸氨基转移酶升高(14.5%)、天冬氨酸氨基转移酶升高(12.0%)、外周水肿(6.0%)和γ-谷氨酰转移酶升高(6.0%)。
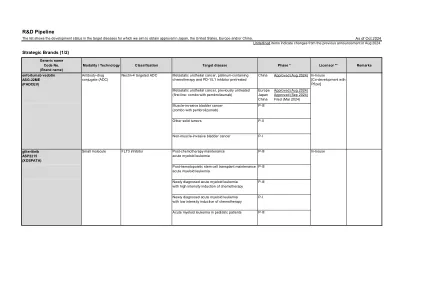
研发管线
与上一公告(2024年8月)的更新:enzalutamide:删除了2024年6月在中国批准用于转移性去势敏感性前列腺癌的描述。enfortumab vedotin:2024年8月在中国获批用于转移性尿路上皮癌,含铂化疗和PD-1/L1抑制剂预处理。2024年8月在欧洲获批用于不可切除或转移性尿路上皮癌的一线治疗(适合含铂化疗)。2024年9月在日本获批用于根治性不可切除的尿路上皮癌的一线治疗。 zolbetuximab:2024 年 10 月在美国获批,2024 年 9 月在欧洲获批,用于一线治疗局部晚期不可切除或转移性 HER2 阴性、claudin 18.2 阳性胃腺癌或胃食管连接部腺癌。avacincaptad pegol:2024 年 10 月在欧洲撤回了用于治疗继发于年龄相关性黄斑变性的地图状萎缩的申请。

2024 年 2 月 5 日|中外制药 FoundationOne CDx 癌症基因组图谱获批用于 PARP 抑制剂 Talazoparib 的伴随诊断,而 Talazoparib 获批用于治疗 BRCA 基因突变阳性且伴有远处转移的去势抵抗性前列腺癌 | 新闻 | 中外制药株式会社
中外制药获批 FoundationOne CDx 癌症基因组图谱可用作 PARP 抑制剂 Talazoparib 的伴随诊断,而 Talazoparib 已获批用于治疗 BRCA 基因突变阳性且伴有远处转移的去势抵抗性前列腺癌


我们如何治疗 HER2 阳性转移性乳腺癌
第一个针对 HER2 受体的药物曲妥珠单抗于 1998 年 9 月获批用于治疗 MBC。2007 年 3 月,拉帕替尼获批,它是一种新型双酪氨酸激酶抑制剂,可同时抑制 HER2 和 EGFR - 表皮生长因子受体。2012 年 6 月,根据 CLEOPATRA 试验,第二种用于治疗 HER2 阳性 MBC 的单克隆抗体帕妥珠单抗获批。3 它与 HER2 受体的不同表位结合,从而防止 HER2 与 HER3 受体发生异二聚化,而曲妥珠单抗则抑制两个 HER2 受体的同二聚化。2013 年 2 月,根据 EMILIA 试验,一种新型抗体-药物偶联物 ado-trastuzumab emtansine (T-DM1) 获批用于治疗 MBC。最近,有三种新药获批用于治疗MBC:2019年12月的曲妥珠单抗德鲁替康(T-DXd)、2020年2月的来那替尼和2020年4月的图卡替尼。值得注意的是,没有证据表明对HER2阴性疾病患者使用HER2靶向治疗,即使免疫组织化学检测HER2评分为2+但荧光原位杂交检测为阴性。
2023 年,100 多种药品和生物制剂的标签将添加儿科信息
• Jardiance(恩格列净)和 Synjardy(恩格列净和盐酸二甲双胍)是获批用于治疗 10 岁及以上儿童患者 2 型糖尿病的新类治疗药物(https://bit.ly/3IdVAkF)。 • Linzess(利那洛肽)是首个获批用于治疗 6 岁及以上儿童患者功能性便秘的药物(https://bit.ly/3UULzQX)。 • Opvee(盐酸纳美芬)是首个获批用于在医疗机构或社区环境中紧急治疗成人和 12 岁及以上儿童患者已知或疑似阿片类药物过量(https://bit.ly/3Td3zEP)的盐酸纳美芬鼻喷雾剂。FDA 还批准了首批非处方阿片类药物过量逆转药物,包括首个仿制盐酸纳洛酮产品,适用于成人和儿童,包括新生儿。这些批准是 FDA 作为其过量预防框架的一部分采取的众多行动之一(https://bit.ly/3POET46)。• Ycanth(斑蝥素)是首个获批用于治疗成人和 2 岁及以上儿科患者的传染性软疣的药物,https://bit.ly/49OFw4B。

新闻稿 Sarclisa 是首个在中国获批的抗 CD38 治疗药物,与标准治疗 VRd 联合使用,用于治疗新诊断的
Olivier Nataf 肿瘤学全球负责人 “四十多年前,赛诺菲进入中国时,就是为了为中国患者带来具有变革性的疗法。此次批准是在 Sarclisa 首次进入中国仅几周后获得的,代表着我们在推进这一使命方面取得了巨大进展。现在,多发性骨髓瘤患者及其提供者可以使用两种新的基于 Sarclisa 的方案,这些方案有可能改善各个疗法的疗效。” 此次批准紧随国家药品监督管理局于 2025 年 1 月初做出的决定,批准 Sarclisa 与泊马度胺和地塞米松 (Pd) 联合用于治疗既往接受过至少一线治疗(包括来那度胺和蛋白酶体抑制剂)的复发或难治性 MM (R/R MM) 成人患者。 除中国之外,在亚太地区,日本目前正在审查 Sarclisa 用于治疗不符合造血干细胞移植 (HSCT) 条件的 NDMM 患者的监管申请。关于 Sarclisa Sarclisa (isatuximab) 是一种 CD38 单克隆抗体,可与 MM 细胞上 CD38 受体上的特定表位结合,诱导明显的抗肿瘤活性。它旨在通过多种作用机制发挥作用,包括程序性肿瘤细胞死亡(凋亡)和免疫调节活性。CD38 在 MM 细胞表面高度均匀地表达,使其成为 Sarclisa 等抗体疗法的靶点。在美国,Sarclisa 的非专有名称是 isatuximab-irfc,以 irfc 为后缀,符合美国食品药品监督管理局发布的非专有生物制品命名行业指南。目前,Sarclisa 已在包括美国、欧盟、日本和中国在内的 50 多个国家/地区获批,涉及多种适应症。根据 ICARIA-MM 的 3 期研究,Sarclisa 已在美国、欧盟和日本获批与 Pd 联合用于治疗既往接受过至少两种疗法(包括来那度胺和蛋白酶体抑制剂)的 R/R MM 患者;在中国,该联合用药也已获批用于既往接受过至少一线疗法(包括来那度胺和蛋白酶体抑制剂)的患者。根据 IKEMA 的 3 期研究,Sarclisa 已在 50 多个国家获批与卡非佐米和地塞米松联合用于治疗,包括在美国用于治疗既往接受过一至三种疗法的 R/R MM 患者,在欧盟用于治疗既往接受过至少
![[转载 - 第 3 批呼叫指南] EE/ELSHI 主题提案指南_2024 年 12 月 17 日 - Google 文档](/simg/9\91d54aead21ed43568e21dd70c1086b54553171d.webp)
[转载 - 第 3 批呼叫指南] EE/ELSHI 主题提案指南_2024 年 12 月 17 日 - Google 文档
其他个人、非金钱方面的参与(其他类型的关系) □ 无(如果“无”,请写“N/A”。)指出任何正在进行的个人关系或有信誉的风险,这些关系或风险可能会产生上述未披露的冲突“表象”(例如,卷入诉讼、研究人员发起的研究、对某一特定领域或主题重要性的个人观点或道德信念,这些观点或信念可能会影响其他人的科学观点,您担任官员、董事、受托人、普通合伙人或员工的组织的活动)。 _______________________________________________________________________________________________________________________________________________________________

GSK 患者援助计划疫苗申请
填写并签署此表。需要患者和医疗保健处方人员的签名。葛兰素史克患者援助计划 (GSK PAP) 不再能够提供单剂量小瓶用于 GSK PAP 补货。在补货发送至站点之前,站点必须在 12 个月内累计共计 10 剂获批产品。在唯一站点地址为所有执业医师批准的剂量将计入累计。如果未能在 12 个月内累计 10 剂获批产品,则补货将被没收。每个唯一站点每年可从 GSK PAP 获得的产品最大数量限制为每种产品 200 剂(20 批 10 支疫苗)。这是一项补货计划。患者应在获得 GSK PAP 批准后接种之前购买的 GSK 疫苗。签署此表,即表示提供者接受该计划的条款和条件,并了解如果在 12 个月内未达到 10 剂获批剂量的增量,将面临风险。