XiaoMi-AI文件搜索系统
World File Search System食品药品


人类基因编辑国际法律治理研究
监管路径 国家或地区 相关法律法规 人类基因编辑监管特点 日本 2000 年《人类克隆技术管制法案》 (The Human 没有制定专门涉及人类胚胎、受精卵、精子 Cloning Regulation Act) ,禁止将克隆人胚胎和 或卵子的伦理指南和法律,其更多依赖于 具有人类和动物遗传物质的胚胎植入子宫。 各个政府部门的监督管理。 2013 年《再生医学安全保障法》 (Regeneration Medicine Promotion Law) ,分级管理再生医疗 风险,科研机构使用基因工程方法修饰后细 胞培养和处理需要通知日本卫生劳动福利部, 获得许可后方可开展研究。 保守 德国 1949 年《德国基本法》 (Basic Law for the Federal 《德国基本法》并没有提供明确和直接的规 Republic of Germany) ,其第 1 条和第 2 条分别规 定,但规定了立法机关必须保护胚胎的基 定了人的尊严、生命权和完整权,保护的范围 本权利。 不仅包括精神病患者、植物人,还包括胎儿和 《胚胎保护法》形成了完全禁止人类胚胎 胚胎。 基因编辑相关临床试验的逻辑森严的刑法 1990 年《胚胎保护法》 (The German Embryo 规制框架。 Protection Law) ,管理人工基因干预生殖系细 胞的情况,其第 5 条第 1 款规定任何人为改变人 类生殖系细胞遗传信息的人,将被处以最高 5 年的监禁或罚款;其第 5 条第 4 款专门规定了非 生殖目的的体外生殖系细胞人工干预不适用第 1 款刑事禁令,确保科研人员在安全性的前提 下进行人类胚胎相关实验的自由。 欧盟 2007 年《欧洲联盟基本权利宪章》 (Charter of 法律允许人类体细胞基因编辑,但明确禁止 Fundamental Rights of the European Union) ,其 在人类胚胎上使用基因编辑技术。 第 3 条禁止基因改造医疗行为,包括人种选择 行为、将人体作为经济收益来源的行为以及克 隆人类行为。 1997 年《人权与生物医学公约》 (Convention on Human Rights and Biomedicine) ,其第 13 条也引 入了对优生学的禁令,规定只能基于预防、诊 断或治疗目的修改人类基因组,并且不允许在 任何后代的基因组中引入任何基因改造。 折衷 美国 2015 年美国白宫发布了有关现阶段反对任何人类 法律不限制技术本身,但限制技术的应用场 种系基因组编辑行为的声明。 2015 年《综合拨 景。鉴于基因编辑是一种工具,不是特定 款法案》 (Consolidated Appropriations Act) ,增 的药物、设备或生物疗法,因而必须在其 加了禁止美国食品药品监督管理局 (Food and 使用的每个领域中审视其是否符合法律 Drug Administration) 使用任何联邦资金资助有 规定。 意修改人类胚胎可遗传物质的研究。 美国食品药品监督管理局禁止涉及可遗传 人类基因组编辑的临床试验,一些州也明 确禁止人类胚胎的特定研究活动。 中国 2020 年《民法典》第 1009 条,从事与人体基因、人 法律对人类体细胞基因编辑的研究和应用不 体胚胎等有关的医学和科研活动,应当遵守法 加以限制,人类胚胎细胞的基因编辑基础 律、行政法规和国家有关规定,不得危害 人体 研究不被禁止,但其临床应用则不被允 健康,不得违背伦理道德,不得损害公共利益。 许,不论是用于生殖目的或是医治患者。 2020 年《刑法》修正案 ( 十一 ) 增加第三百三十 六条,将基因编辑、克隆的人类胚胎植入人体 或者动物体内,或者将基因编辑、克隆的动物 胚胎植入人体内,情节严重的,处三年以下有 期徒刑或者拘役,并处罚金;情节特别严重的, 处三年以上七年以下有期徒刑,并处罚金。

消化
• 网络安全:《联邦食品药品监督管理局法案》第 3305 条要求在“网络设备”的上市前申请中纳入网络安全信息,并规定不遵守新的网络安全要求是《联邦食品药品监督管理局法案》禁止的行为。术语“网络设备”是指 (1) 包含由发起人验证、安装或授权作为设备或设备中的软件的设备;(2) 具有连接到互联网的能力;以及 (3) 包含由发起人验证、安装或授权的任何可能容易受到网络安全威胁的技术特征的设备。总之,网络设备的发起人必须提供计划来监控、识别和解决任何上市后网络安全漏洞;建立和维护程序以确保设备和相关系统的网络安全;提供软件物料清单;并满足 FDA 可能制定的任何其他要求,以确保设备和相关系统的网络安全。FDA 可能会免除某些设备或设备类别的这些网络安全要求。

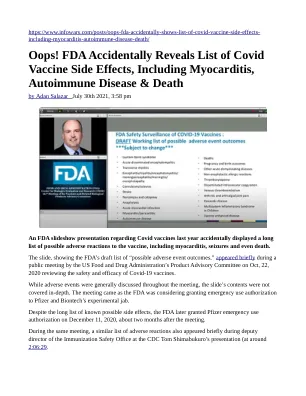
糟糕!FDA 意外披露新冠疫苗副作用清单,包括心肌炎、自身免疫性疾病和死亡
这张幻灯片展示了 FDA 的“可能的不良事件结果”草案清单,在美国食品药品管理局产品咨询委员会于 2020 年 10 月 22 日举行的公开会议上短暂出现,该会议旨在审查新冠疫苗的安全性和有效性。

adb-brief-300-疫苗制造-孟加拉国。......
1 EUA,即紧急使用清单,是全球组织和国家监管机构(例如世界卫生组织 (WHO) 或美国食品药品监督管理局 (FDA))使用的基于风险的工具或程序,用于评估和列出未经许可的疫苗、治疗药物和体外诊断产品,最终目的是加快向受公共卫生紧急事件(包括传染病)影响的人们提供这些产品。这些工具可帮助感兴趣的采购机构和政府根据一组基本的现有质量、安全性和有效性以及性能数据来确定使用特定产品的可接受性。2 美国食品药品监督管理局。2021 年。FDA 批准首款 COVID-19 疫苗。新闻稿。8 月 23 日。3 N. Gozzi 等人。2023 年。评估 COVID-19 疫苗不平等的影响:一项建模研究。自然通讯。14。3272。4 CA Kunyenje 等人。 2023. 非洲低收入国家 COVID-19 疫苗不平等问题。公共卫生前沿。11:1087662。

besc;主持人的脚本;模块3。
s Lide S 7至9呈现表3的缩短版本,手套类型和适应症,从牙科医疗保健环境中的感染指南(2003年)。列出了三种类型的手套:患者检查手套,外科医生的手套和非医疗手套。 患者检查手套用于患者护理,检查,其他非手术程序,涉及与粘膜接触和实验室程序。 它们是由美国食品药品监督管理局(FDA)调节的医疗设备,可作为非遗产和无菌的一次性一次性物品。 仅将它们用于一名患者并适当丢弃。列出了三种类型的手套:患者检查手套,外科医生的手套和非医疗手套。患者检查手套用于患者护理,检查,其他非手术程序,涉及与粘膜接触和实验室程序。它们是由美国食品药品监督管理局(FDA)调节的医疗设备,可作为非遗产和无菌的一次性一次性物品。仅将它们用于一名患者并适当丢弃。

ANDA 提交——根据 GDUFA 对简化新药申请进行修订
I. 引言 本指南旨在向申请人解释 2022 年仿制药用户费用修正案 (GDUFA III)2 中设立的评估目标如何适用于根据《联邦食品药品和化妆品法案》(FD&C Act)第 505(j) 节(21 USC 355(j))3 提交给食品药品管理局的简化新药申请 (ANDA) 或事先批准补充文件 (PAS) 的修正案。本指南介绍了修正案的分类和类别,并解释了修正案提交如何影响申请的评估目标日期。本指南取代了 2018 年 7 月的行业 ANDA 提交指南——根据 GDUFA 对简化新药申请的修正案。一般而言,FDA 的指导文件并不规定具有法律强制力的责任。相反,指南描述了该机构目前对某一主题的想法,除非引用特定的监管或法定要求,否则应仅将其视为建议。机构指南中使用“应该”一词的意思是建议或推荐某事,但不是要求。

更新后的 COVID 疫苗代码 - 2023 年 6 月 10 日
根据美国食品药品管理局 (FDA) 现有的紧急使用授权 (EUA) 或生物制品许可申请 (BLA) 的规定,疫苗接种已关闭。自 2023 年 9 月 12 日起,19 岁以下会员的 COVID-19 疫苗现已纳入儿童疫苗 (VFC) 计划。已加入

新疗法显示出对致命神经疾病的有望:研究
最终,如果它证明了这一点,Agar表示,他希望它可以成为Biogen的Qalsody的共同治疗,Biogen的Qalsody是一种突破性的方案,该方案在2023年获得了食品药品监督管理局的加速认可,该疗法通过减少SOD1基因的数量来生产该人体产生的SOD1基因的数量。