XiaoMi-AI文件搜索系统
World File Search System核酸酶

抗抑性RT-qPCR预混液
储存和稳定性: 抗抑性 RT-qPCR 预混液采用干冰 / 蓝冰运输。到货后储存于 -20°C 下,以获得最佳稳定性。应避免反复 冻融循环。运输过程中解冻不影响产品性能。每次解冻后应混合 / 平衡溶液以避免分相。 有效期: 在外包装盒标签上的有效期内,在推荐条件下储存并正确处理时,试剂盒可保持完整活性。 安全预防措施: 处理试剂前请阅读并理解 SDS (安全数据表)。首次发货时提供 SDS 的纸质版文件,此后可应要求提 供。 质量控制: Meridian 遵守 ISO 13485 质量管理体系运行。抗抑性 RT-qPCR 预混液及其组分在活性、持续合成能 力、效率、热激活、灵敏度、无核酸酶污染和无核酸污染等方面均经过广泛测试 注: 仅供科研和 / 或进一步生产使用。

如何引用:Mariano Junior CG,Oliveira VC,AmbrósioCE。小动物中的基因编辑用于转化医学:评论。动画复制。 2
与其他工程核酸酶(例如锌指核酸酶(ZFN)和类似转录激活剂样效应子核酸酶(Talens))相比,CRISPR/CAS9系统是一种更简单,更通用的方法,自从发现其CRISPR基因组编辑效率以来,基于CRISPR的基因组效率就可以使多种和不同类型的编辑效率提高。这些基因编辑的进步通过比以往任何时候都更加简单和有效性来启用精确的基因组编辑,从而彻底改变了生物技术。该工具已成功应用于各种动物物种,包括牛,猪,狗和其他小动物。工程核酸酶切断了特定靶位置的基因组,触发了细胞修复损伤的机制,并将突变引入特定的基因组位点。本评论讨论了基于基因组的新型CRISPR/CAS9编辑工具,为提高效率和特异性而开发的方法,在动物模型和转化医学上使用基因编辑的方法以及基于CRISPR的基因编辑方法的主要挑战和局限性。

利用双主键编辑对大型 DNA 序列进行可编程删除、替换、整合和反转
与疾病相关的人类遗传变异范围从单碱基对替换到兆碱基重复、缺失和重排 1-3 。可以在人类细胞中安装、纠正或补充这些致病变异的基因编辑方法有可能促进对遗传疾病的了解,也可能实现新的治疗方法 4、5。过去十年来,已经开发出几种基于 CRISPR-Cas 系统的哺乳动物细胞基因编辑方法 6,包括核酸酶 7-9 、碱基编辑器 10、11 和主要编辑器 12 ,每种方法都有可能解决一组已知的致病序列变化。CRISPR-Cas 核酸酶(如 Cas9)可用于通过创建导致不受控制的插入/缺失混合的 DSB 来破坏基因。此外,配对的 Cas9 核酸酶策略可以介导长度从约 50 到 > 100,000 个碱基对的基因组 DNA 序列的靶向删除 13 。通过提供线性供体 DNA 序列,可以通过末端连接或同源性定向修复 (HDR) 过程在单个切割位点或成对切割位点之间定向插入新的 DNA 序列 14, 15。单核酸酶和成对核酸酶编辑方法虽然用途广泛,但它们也存在相当大的缺点。DNA 供体敲入伴随着高效的 indel 副产物 16,因为在大多数细胞类型中,HDR 与末端连接过程相比通常效率低下 17, 18。使用成对核酸酶进行靶向删除会产生多种副产物 13, 19,而且缺失的精确位置受到 PAM 可用性的限制。此外,在靶位或脱靶位点的 DSB 可促进大面积缺失 20-22、染色体异常 23、24 和染色体碎裂 25。 DSB 倾向于生成不良副产物和染色体改变的复杂混合物 26 - 28,这在应用基于核酸酶的编辑来操作较大的 DNA 序列时带来了相当大的挑战,特别是在治疗环境中。

基因组/基因编辑工具的观点
摘要:从理论上讲,可以区分等于或超过16 bp的DNA序列的DNA序列特异性识别蛋白可能是哺乳动物基因组独有的。长期序列的核酸酶,例如天然存在的归巢核酸酶和人工设计的ZFN,TALEN和CAS9-SGRNA。与其他对应物(通过蛋白质部分识别DNA靶位点的其他对应物相比,CAS9使用单个指南RNA(SGRNA)作为DNA靶标识别的模板。由于设计和合成目标位点的SGRNA的简单性,CAS9-SGRNA已成为基因组编辑的最新工具。此外,Cas9-SgrNA的RNA引导的DNA识别活性与HNH结构域和RUVC结构域的非平均链中的核酸酶活性无关,而HNH核酸酶无核酶无效无效无效活性无效(H 840 A)和RUVC核酸酶核酸酶活性无效null null突变(识别10 A)。与SGRNA,CAS9,Cas9(D 10 A),Cas9(H 840 A)和Cas9(D 10 A,H 840 A)一起用于实现双重链断裂,互补的链断管破裂,非满足链破裂,并且分别在TARPEC上进行破裂。基于这种独特的特征,可以在靶位点内或周围引入许多工程酶活性,例如DNA甲基化,组蛋白甲基化,组蛋白乙酰化,胞苷脱氨酸,腺嘌呤脱氨基和启动引导突变。为了防止Cas9衍生物的持久表达靶向,开发了许多瞬态表达方法,包括直接递送Cas9-SgrNA核糖蛋白。生物安全问题在体内应用中是必不可少的;已经设计了包装到病毒样颗粒或细胞外囊泡中的CAS9-SGRNA,已经报道了一些体内治疗试验。

CRISPR/Cas9 敲除质粒
CRISPR/Cas 系统是一种适应性免疫防御机制,古细菌和细菌利用该系统降解外来遗传物质。在这些生物体中,噬菌体的外来遗传物质被获取并整合到 CRISPR 基因座中 (1,2)。这种新物质也称为间隔物,可产生序列特异性片段,用于未来抵抗噬菌体感染。这些序列特异性片段被翻译成短 CRISPR RNA (crRNA),并通过 CRISPR 相关 (Cas) 蛋白的核酸酶活性引导互补入侵 DNA 的切割,该蛋白也由 CRISPR 基因座编码 (1,2)。II 型 CRISPR 系统的 Cas9 核酸酶具有 RNA 结合域、α 螺旋识别叶 (REC)、包括用于 DNA 切割的 RuvC 和 HNH 的核酸酶叶以及原间隔物相邻基序 (PAM) 相互作用位点 (1,2)。 crRNA 通过与 REC 叶内的桥螺旋结合与 Cas9 核酸酶形成复合物,并与 crRNA 的骨架形成多个盐桥 (1,2,3)。

活细胞成像揭示了 Cas9 和 Cas12a 靶标搜索灵活性与效率之间的权衡
摘要 CRISPR-Cas 系统已被广泛用作基因组编辑工具,其中两种常用的 Cas 核酸酶是 Spy Cas9 和 Lb Cas12a。虽然这两种核酸酶都使用 RNA 向导来寻找和切割靶 DNA 位点,但这两种酶在原间隔区相邻基序 (PAM) 要求、向导结构和切割机制方面有所不同。在过去的几年里,合理工程设计导致了 PAM 放宽变体 Sp RYCas9 和 imp Lb Cas12a 的诞生,以拓宽可靶向的 DNA 空间。通过使用它们的催化无活性变体 (dCas9/dCas12a),我们量化了蛋白质特异性特征如何影响靶标搜索过程。为了进行量化,我们将这些核酸酶与光激活荧光蛋白 PAmCherry2.1 融合,并在大肠杆菌细胞中进行单粒子追踪。通过跟踪分析,我们推导出了每种具有非靶向 RNA 向导的核酸酶的动力学参数,这强烈表明 Lb dCas12a 变体对 DNA 的询问比 Spy dCas9 更快。在存在靶向 RNA 向导的情况下,模拟和细胞成像均证实 Lb dCas12a 变体在找到特定靶位点方面更快、更高效。我们的工作展示了使用强大的框架工作放宽 Spy dCas9 和 Lb dCas12a 中的 PAM 要求的权衡,这可以应用于其他核酸酶以量化它们的 DNA 靶标搜索。

远程随机电路中的运输和纠缠增长
具有可编程核酸酶的基因组编辑对临床翻译表现出了巨大的希望,但也揭示了由染色体易位引起的遗传毒性的风险或在脱靶位点插入突变的插入。在这里,我们描述了一种创新的测定法,以识别和量化源自CRISPR-CAS核酸酶或Talens的靶向活性的染色体畸变。cast-seq还检测了新型的染色体重排类型,包括同源性重组介导的同源介导的易位。取决于使用的设计师核酸酶,易位发生在0-0.5%的基因编辑的人类干细胞和约20%的靶基因座中含有大差点。总而言之,铸造SEQ分析与干细胞的治疗编辑特别相关,以在基因编辑产物的临床应用之前进行彻底的风险评估。

基础编辑:进步和治疗机会
要用作治疗性,基因组编辑工具必须表现出较高的靶向效率和最小的有害或不需要的脱靶编辑,并且可传递到感兴趣的器官。重要的是要注意,疾病靶标将决定必须满足这些标准的确切程度。在该领域的早期努力使用了诸如锌指核酸酶和转录激活剂样效应子核酸酶(TALENS)之类的平台,但是对于每个新的目标编辑站点8设计和验证了新的锌指核酸酶或塔伦蛋白的要求,这些方法受到了阻碍。然而,这些广泛的蛋白质重新设计需求通过发现,机械阐明和适应性的适应性来缓解,以簇定期间隔短的短质体重复(CRISPR)平台进行基因组编辑。

通过 DNA 修复调节和 pegRNA 工程改进基于核酸酶的引物编辑 Panagiotis Antoniou* 1、Louis Dacquay* 1,2、Niklas Selfjord 1
通过 DNA 修复调节和 pegRNA 工程改进基于核酸酶的引物编辑 Panagiotis Antoniou* 1 、Louis Dacquay* 1,2 、Niklas Selfjord 1 、Katja Madeyski-Bengtson 3 、Anna-Lena Loyd 3 、Euan Gordon 4 、George Thom 5 、Pei-Pei Hsieh 1 、Sandra Wimberger 1 、Saša Šviković 1 、Mike Firth 6 、Nina Akrap 1 、Marcello Maresca 1# 和 Martin Peterka 1# 1 基因组工程,Discovery Sciences,研发部,阿斯利康,瑞典哥德堡 2 Promega Corporation,美国威斯康星州麦迪逊 3 转化基因组学,Discovery Sciences,研发部,阿斯利康,瑞典哥德堡 4 发现生物学,Discovery Sciences,研发部,阿斯利康,瑞典哥德堡 5 英国剑桥阿斯利康公司发现科学研发部体内表达生物制剂 6 英国剑桥阿斯利康公司发现科学研发部数据科学与定量生物学 * 这些作者的贡献相同 # 通信地址:marcello.maresca@astrazeneca.com martin.peterka@astrazeneca.com
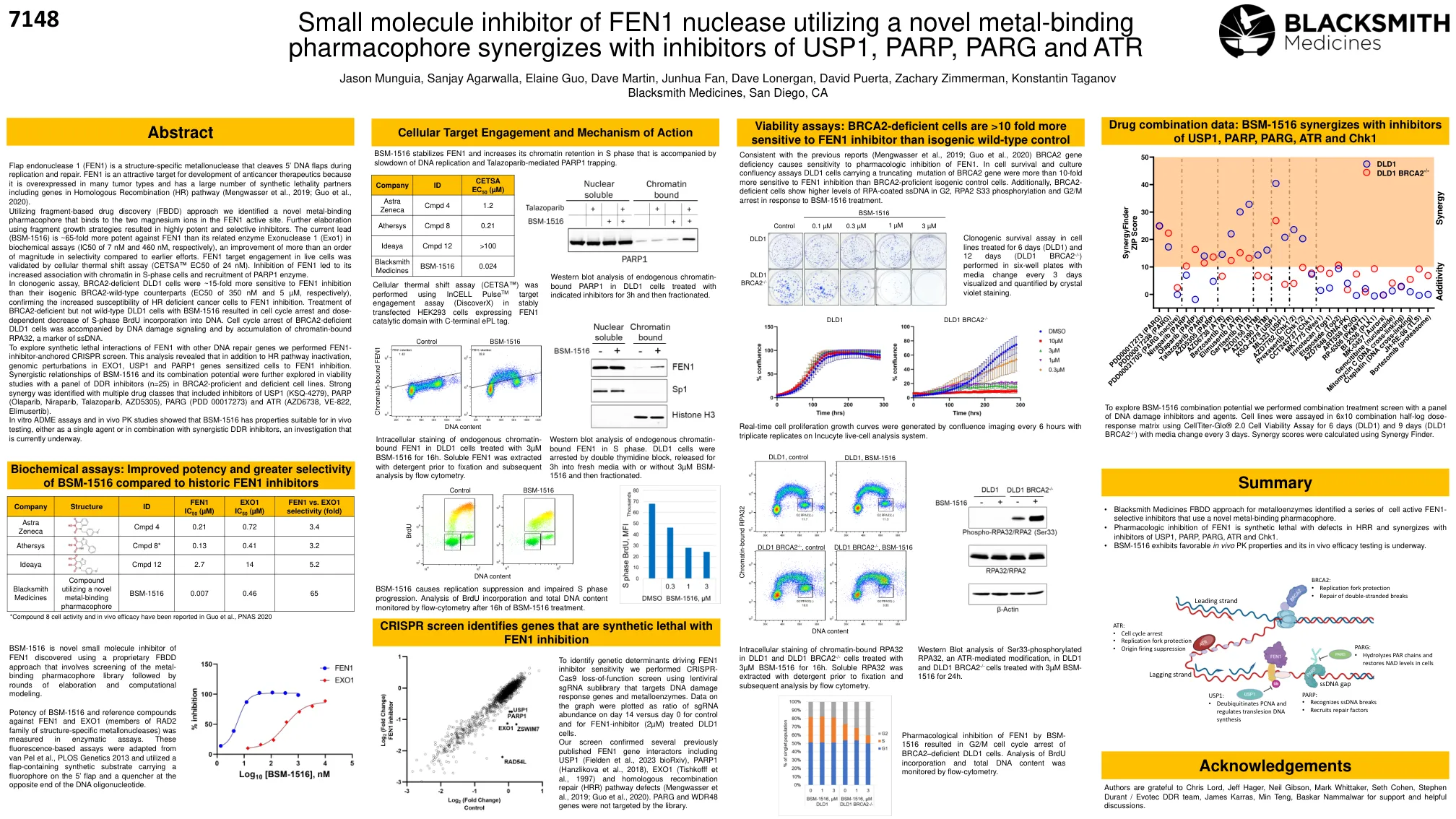
使用新型金属结合药效团与USP1,PARP,PARG和ATR抑制剂协同的Fen1核酸酶的小分子抑制剂
皮瓣核酸内切酶1(Fen1)是一种结构特异性的金属核酸酶,在复制和修复过程中切割5'DNA瓣。fen1是开发抗癌疗法的有吸引力的靶标,因为它在许多肿瘤类型中过表达,并且具有大量的合成致死性伴侣,包括同源重组基因(HR)途径(Mengwasser等,2019; Guo等,2020)。利用基于碎片的药物发现(FBDD)方法,我们确定了一种新型的金属结合药效团,该药效团与Fen1活性位点中的两个镁离子结合。使用碎片增长策略进一步阐述导致高度有效和选择性抑制剂。在生物化学测定中(分别为7 nm和460 nm的IC50),对FEN1的当前铅(BSM-1516)对FEN1的有效性比其相关酶外核酸酶1(EXO1)高65倍,与早期努力相比,改善了量的更大范围。fen1靶标在活细胞中的靶标参与通过细胞热偏移分析验证(CETSA