XiaoMi-AI文件搜索系统
World File Search System汇集的

犬乳腺肿瘤类器官活体生物库可作为人类乳腺癌的比较模型
犬乳腺肿瘤具有作为转化肿瘤学中自然发生的乳腺癌模型的巨大潜力,因为它们与人类乳腺肿瘤具有相同的环境风险因素、关键组织学特征、激素受体表达模式、预后因素和遗传特征。我们旨在开发允许对犬乳腺肿瘤 (CMT) 进行功能分析的体外工具,因为我们对驱动这些异质性肿瘤生长的潜在生物学了解甚少。我们建立了来自 16 名患者的 24 个类器官系的长期培养,包括来自正常乳腺上皮或良性病变的类器官。CMT 类器官重现了它们所来自的原发组织的关键形态学和免疫组织学特征,包括激素受体状态。此外,遗传特征(驱动基因突变、DNA 拷贝数变异和单核苷酸变异)在肿瘤-类器官对中得到保留。我们展示了 CMT 类器官如何成为体外药物测定的合适模型,并可用于研究特定突变是否可预测治疗结果。此外,我们可以对 CMT 类器官进行基因改造,并使用它们进行汇集的 CRISPR/Cas9 筛选,其中文库表示得到准确维护。总之,我们提出了一个强大的 3D 体外临床前模型,可用于转化研究,其中可以从同一患者体内繁殖来自正常、良性和恶性乳腺组织的类器官,以研究肿瘤发生。

全基因组 CRISPR 筛选揭示了家蚕细胞活力和抗非生物和生物胁迫所必需的基因
高通量基因筛选是一种强大的方法,可用于在全基因组范围内研究基因功能并识别对某些压力负责的基因。在这里,我们开发了一种 piggyBac 策略,可将汇集的 sgRNA 文库稳定地递送到细胞系中。我们使用这种策略在家蚕细胞中进行基于全基因组成簇的规律间隔短回文重复技术 (CRISPR)-Cas9 的筛选。我们首先构建了一个包含 94,000 个 sgRNA 的单向导 RNA (sgRNA) 文库,该文库靶向 16,571 个蛋白质编码基因。然后,我们使用 piggyBac 转座子在 BmE 细胞中生成敲除集合。我们确定了 1006 个在正常生长条件下对细胞生存至关重要的基因。在已确定的基因中,82.4%(829 个基因)与七种动物物种中的必需基因同源。我们还确定了 838 个基因,它们的缺失促进了细胞生长。接下来,我们分别使用温度和杆状病毒对生物或非生物胁迫进行了针对特定环境的阳性筛选,从每个筛选中确定了几个关键基因和途径。总之,我们的结果为家蚕基因组的功能注释和解释导致各种条件的关键基因提供了一个新颖而通用的平台。这项研究还展示了在非模式生物中进行全基因组 CRISPR 筛选的有效性、实用性和便利性。

SBAA3018信息管理
单元1简介数据,信息,情报,信息技术,信息系统,进化,基于功能和层次结构,系统开发方法,功能信息系统,DSS,EIS,KMS,KMS,GIS,GIS,GIS,国际信息系统。数据:数据是当前未用于决策目的的事实。例如,薪资记录,应收帐款数据,人员数据等。“数据是指通常是由于经验,观察或实验或计算机系统中的过程或一组前提而收集的事实的集合。”数据可能包括数字,单词或图像,特别是作为一组变量的测量或观察结果。数据通常被视为从中得出信息和知识的最低抽象水平。根据Ackoff的说法,“数据是代表对象,事件及其环境的属性的符号。它们是观察产物。观察是感官。当然,传感仪器的技术是高度发达的。”信息是数据通过关系连接的含义。此“含义”可能很有用,但不一定是。在计算机句子中,一个关系数据库从存储在其中的数据中获取信息。信息来自数据,可用于解决问题。信息,因此是数据的潜在函数。一旦数据被筛选并组织起来,以使其与当前的决策背景相关,则可以称为信息。准确性7。有关信息组织数据数据的重要点,具有上下文数据,具有与知识的关系派的上下文数据,并存储了知识沟通的知识汇集的物理表示,或接受信息特征的知识动作,以了解信息的特征1。及时性6。适当性2。简洁3。频率8。可理解性4。相关9。完成

GREPore-seq:通过长距离 PCR 和纳米孔测序检测基因编辑后变化的强大工作流程
为了充分发挥基因编辑技术在临床治疗中的巨大潜力,需要彻底评估靶向编辑和非预期编辑的后果。然而,目前缺乏一种全面、流水线化、大规模且经济的工作流程来检测基因组编辑结果,特别是插入或删除大片段。在这里,我们描述了一种通过对条形码长距离 PCR 产物进行纳米孔池测序来有效准确地检测 CRISPR-Cas9 编辑后的多个基因变化的方法。为了克服纳米孔测序的高错误率和插入缺失,我们开发了一种流程,通过对纳米孔扩增子测序 (GREPore-seq) 的读取进行 grepping 来捕获条形码序列。GREPore-seq 可以检测 NHEJ 介导的双链寡脱氧核苷酸 (dsODN) 插入,其准确度与 Illumina 下一代测序 (NGS) 相当。GREPore-seq 还可以识别 HDR 介导的大基因敲入,这与 FACS 分析数据高度相关。还检测到了 HDR 编辑后的低水平质粒骨架插入。我们建立了一个实用的工作流程来识别遗传变化,包括量化 dsODN 插入、敲入、质粒骨架插入和 CRISPR 编辑后的大片段缺失。该工具包用于对汇集的长扩增子进行纳米孔测序,在评估靶向 HDR 编辑和超过 1 kb 的意外大插入缺失方面应具有广泛的应用。GREPore-seq 可在 GitHub 上免费获取(https://github.com/lisiang/GREPore-seq)。

俄罗斯代表形容词构造的二元组的定量方法
natalia bobkova - Clle,CNRS和ToulouseUniversitéde toulouseJaurèsFabioMonterni - Clle,CNRS和ToulouseJeanJaurès大学摘要本文致力于罗素(Russian)派生形容词中的后缀之间的竞争。,它提出了基于俄罗斯国家语料库的大规模定量分析。它的主要目标是为确定确定这些衍生物中后缀选择的属性做出贡献。俄罗斯的代名词形容词派生使用了各种各样的指数。其中大多数是三个主要后缀的语音变体(扩展)-n-, - SK-和-OV-。后者可以被认为是基本的,构成了我们分析的重点。为这项研究构建了两个数据集,其中一个包含上面的后缀之一,一个更具体的包含Doublet的数据集,即形容词在同一基础上具有不同后缀。通过各种统计模型分析了两组的数据。我们的结果在全球范围内提供了对先前文献中先前进行的观测值的定量确认。特别是,我们表明-n-在俄罗斯的衍生系统中占有特定的位置,因为它的生产力较低,其衍生物倾向于较不透明,并且更容易显示词汇化的含义,这些含义指向朝向定性 - 相关语义谱的定性极点。- SK-和 - OV-更可能形成双重峰(附着在相同的基础上),这是一个进一步的论点,支持它们之间更大的同质性,而不是-n-。关键字:俄语,衍生形态,代表形容词,定量语言学,语料语言学,统计方法的统计方法1.引入俄罗斯名词中形容词的推导是观察和分析词缀竞争的有趣基础。代名词形容词(我们可能在全球范围内将其表征为具有关系价值)实际上可能是通过这种语言来得出的,主要是通过三种不同的后缀 - n-, - sk-和 - ov-或多个变体,基本上扩展了后者的变体。文献中已经进行了几次尝试,以隔离确定选择一个或其他后缀 /变体的因素,以绝对或倾向(参见< / div>)Townsend 1975; Švedova1980; Zemskaja 2015; Hénault&Sakhno 2016等)。 确定的因素包括基本名词和衍生物的语音,形态学,语义和词源特性,或与两者之间关系相关的特性。 然而,除了对小词汇集的一些研究外,仍然缺乏对这种现象的观察。 我们在本文中进行的研究是对俄罗斯代表形容词进行大规模分析的方向的第一步。 我们的主要目标是构建强大的统计模型,以预测相关派生中的后缀的选择。 特别是,我们从俄罗斯国家语料库中构建了两个不同的数据集:一般形容词的一般数据集,其中包含上面列出的主要后缀之一,以及在语料库中遇到的所有Doublet的数据集,即>Townsend 1975; Švedova1980; Zemskaja 2015; Hénault&Sakhno 2016等)。确定的因素包括基本名词和衍生物的语音,形态学,语义和词源特性,或与两者之间关系相关的特性。然而,除了对小词汇集的一些研究外,仍然缺乏对这种现象的观察。我们在本文中进行的研究是对俄罗斯代表形容词进行大规模分析的方向的第一步。我们的主要目标是构建强大的统计模型,以预测相关派生中的后缀的选择。特别是,我们从俄罗斯国家语料库中构建了两个不同的数据集:一般形容词的一般数据集,其中包含上面列出的主要后缀之一,以及在语料库中遇到的所有Doublet的数据集,即形容词在同一底座上用不同的后缀构建。实际上我们将表明的是,对双重组的研究可能会阐明系统的全球动态,特别是当这样的

用子...
昆虫学采样和存储条件通常会优先考虑效率,实用性和形态特征的保守性,因此可能是DNA保存的次优。这种做法可能会影响下游分子应用,例如高通量基因组文库的结构,这通常需要大量的DNA输入量。在这里,我们使用了实用的TN5转座酶标记的基于基于96孔板的库制备方法,并从昆虫腿的低屈服DNA提取物中进行了优化,这些昆虫的DNA提取物是在亚最佳条件下存储的DNA保存的。将样品在野外车辆中长时间保存,然后在冰箱中的乙醇中长期存储或在室温下干燥。通过将DNA输入减少到6ng,可以处理更多具有亚最佳DNA产量的样品。我们将这种低DNA输入与市售标记酶的6倍稀释匹配,从而大大降低了库制备成本。通过直接放大后单个图书馆汇集的成本和工作量进一步抑制。我们生成了90个样本中88个中等覆盖范围(> 3倍)基因组,平均覆盖率约为10倍。与储存在乙醇中的样品相比,与储存的样品相比,DNA的DNA明显较少,但这些样品具有较高的测序统计量,其测序读数较长,内源性DNA的速率更高。此外,我们发现基于标记的库制剂的效率可以通过彻底的放大后珠子清理来提高,该珠子可以选择不针对短和大的DNA片段。通过打开使用亚最佳保存的低产量DNA提取物的机会,我们扩大了昆虫标本的整个基因组研究的范围。因此,我们期望这些结果和该协议对于昆虫学领域的一系列应用都有价值。

CRISPR 筛选是否能提供下一代治疗靶点? Francisca Vazquez 1 和 William R. Sellers 1,2,3
精准癌症医学的成功取决于我们能否发现并针对患者肿瘤中的特定弱点进行治疗。理想情况下,我们将确定人类癌症中的所有靶点弱点,即维持癌症活力的靶点组合,并开发药物来抑制每个靶点。最后,我们需要非交叉耐药性治疗组合来克服人类肿瘤中发现的潜在亚克隆异质性。我们距离这一目标还很远,因为我们不完全了解癌症的弱点,而且我们缺乏针对大多数已识别弱点的药物。现在有两项进展使得在识别所有癌症弱点方面取得更大进展成为可能。首先,大量癌细胞模型的可用性和特性,虽然这些模型仍不完整,但可以开始模拟人类癌症的多样性。在这里,癌细胞系百科全书显著改变了在大量高度表征的细胞系 (1-3) 中分析治疗活性的能力。该集合现在已注释了包含 1,700 多个细胞系的数据集(可在 depmap.org 上获取)。模型生成方面的最新进展应该可以大大扩展这种多样性。第二次革命是在没有小分子抑制剂的情况下诱导基因特异性功能丧失的能力。此外,随着基因组规模 CRISPR 筛选的出现,现在可以以汇集的形式有效地改变每个基因的功能。CRISPR 筛选现在在大量细胞系或同源细胞系对中体外常规进行,在同基因小鼠模型中体内常规进行(4-6)。这些方法已经确定了正在药物发现计划中或新抑制剂处于临床开发中的治疗靶点(图 1)。也许同样重要的是现在能够确定特定疗法的更广泛有效性或无效性

圣费尔南多地震会议
本出版物中讨论的任何信息、设备、产品或工艺的完整性、适用性或实用性,不承担任何责任。未首先就其是否适用于任何一般或特定应用获得专业建议,不得使用此信息。任何使用此信息的人都应对此使用承担所有责任,包括但不限于侵犯任何专利。引用此出版物为:Davis, C. A., K. Yu, and E. Taciroglu (2021)。圣费尔南多地震会议 - 生命线工程 50 年:摘要集,Lifelines2021-22,加州大学洛杉矶分校自然灾害风险与恢复力中心报告编号。GIRS-2021-05,第 1 版,2021 年 3 月 22 日,doi:10.34948/N3QP4X,https://doi.org/10.34948/N3QP4X。摘要 自 1971 年加州洛杉矶圣费尔南多地震后生命线地震工程成为主要实践领域以来,该领域已取得 50 年的进步。在此期间,生命线工程已在世界各地应用,并扩展到应对地震以外的其他灾害。生命线工程现在被认为是确保社区抵御任何和所有灾害的关键方面。因此,在 1971 年圣费尔南多地震 50 周年之际,认识到过去半个世纪生命线工程的进步非常重要。要理解、改进和实施生命线基础设施系统的灾害恢复能力,我们必须认识到我们在实践中处于什么位置、我们如何到达这里以及我们应该去哪里。我们还必须认识到它对于提高社区恢复能力的重要性日益增加。2021-22 年生命线会议汇集了从业者、研究人员、教育工作者、材料供应商、创新者、服务用户和其他与改善生命线基础设施系统相关的专家。本卷中汇集的摘要代表了实践现状和最新技术,并描述了生命线基础设施系统的未来需求。

2020-MKO-Process-Safety-Symposium-Proceedings.pdf
我很高兴欢迎您参加 Mary Kay O’Connor 过程安全研讨会论文集 2020 年年度研讨会。2020 年研讨会是该系列的第 23 届,由于正在发生的冠状病毒大流行的影响,首次以虚拟方式举行。我们的研讨会是为了纪念我们的同名人 Mary Kay O’Connor 和我们的创始主任 M. Sam Mannan 教授。研讨会是一项重要的年度活动,重点关注影响过程安全和风险管理的研究、教育、培训和服务问题。非常感谢您的参与,特别是在这个艰难时期,我们都在努力应对冠状病毒大流行对我们的生活和日常运营的影响。您的参与对于研讨会的成功以及推动过程安全技术和概念的事业以使行业更加安全至关重要。我们认为,积极改进工艺安全计划对企业有利,尤其在当前困难时期,对行业利润有积极影响。举办本次年度研讨会的目的有三点。首先,本次年度活动提供了一个独立、公正的论坛,供业界、学术界、政府机构和其他利益相关者交流思想和进行讨论,共同探讨工艺安全领域的关键研究问题和进展。其次,它提供了一个绝佳的交流平台,工艺安全专业人员可以在此建立点对点联系,以备将来之需,同时了解他们可以从他人那里获得的各种服务。最后,我们坚信,在我们今天应对 COVID-19 的不确定局面时,良好、扎实的研究可以帮助解决当今行业面临的复杂而有趣的问题。识别这些问题并利用研讨会讨论中汇集的专业知识交换想法和意见将为解决当前问题提供背景信息。此外,研讨会的参与者还可以借此机会了解 Mary Kay O'Connor 过程安全中心所做的前沿研究。这些会议记录包含研讨会计划、在研讨会上发表并在截止日期前提交的论文以及中心的其他信息内容。我们希望您从本次研讨会中获得最大收益,并强烈鼓励您参加虚拟讨论。我们期待着 2021 年 10 月在德克萨斯 A&M 大学举行的面对面研讨会上欢迎大家。请随时与我或其他中心人员联系,提供您对研讨会和中心其他活动的想法和意见。祝愿一切平安恢复正常。
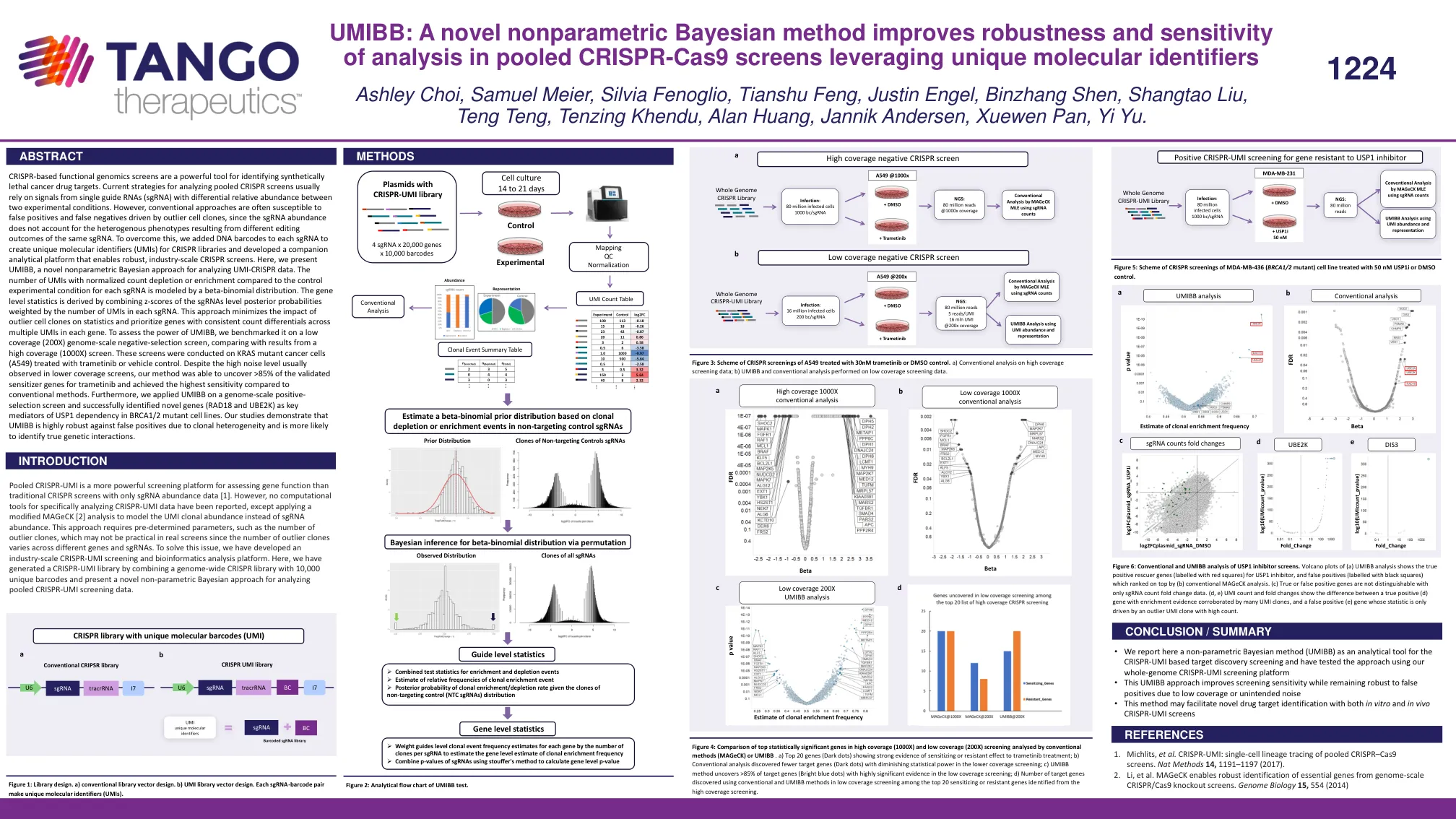
UMIBB:一种新的非参数贝叶斯方法提高了稳健性和灵敏度
基于 CRISPR 的功能基因组学筛选是识别合成致死癌症药物靶点的有力工具。目前分析汇集的 CRISPR 筛选的策略通常依赖于来自在两种实验条件下具有不同相对丰度的单个向导 RNA (sgRNA) 的信号。然而,传统方法通常容易受到由异常细胞克隆驱动的假阳性和假阴性的影响,因为 sgRNA 丰度不能解释由相同 sgRNA 的不同编辑结果导致的异质表型。为了克服这个问题,我们在每个 sgRNA 中添加了 DNA 条形码,以创建 CRISPR 文库的唯一分子标识符 (UMI),并开发了一个配套的分析平台,以实现强大的工业规模 CRISPR 筛选。在这里,我们介绍了 UMIBB,一种用于分析 UMI-CRISPR 数据的新型非参数贝叶斯方法。与每个 sgRNA 的对照实验条件相比,具有标准化计数消耗或富集的 UMI 数量由 beta-二项分布建模。基因水平统计数据是通过将 sgRNA 水平后验概率的 z 分数与每个 sgRNA 中 UMI 的数量加权而得出的。这种方法最大限度地减少了异常细胞克隆对统计数据的影响,并优先考虑每个基因中多个 UMI 之间计数差异一致的基因。为了评估 UMIBB 的功效,我们在低覆盖率(200X)基因组规模负选择筛选上对其进行了基准测试,并与高覆盖率(1000X)筛选的结果进行了比较。这些筛选是在用曲美替尼或载体对照处理的 KRAS 突变癌细胞(A549)上进行的。尽管在较低覆盖率筛选中通常会观察到高噪音水平,但我们的方法能够发现 >85% 的曲美替尼已验证的致敏基因,并且与传统方法相比实现了最高的灵敏度。此外,我们将 UMIBB 应用于基因组规模的正向选择筛选,并成功确定了新基因(RAD18 和 UBE2K)是 BRCA1/2 突变细胞系中 USP1 依赖性的关键介质。我们的研究表明,UMIBB 对克隆异质性导致的假阳性具有很高的稳健性,并且更有可能识别真正的遗传相互作用。