XiaoMi-AI文件搜索系统
World File Search SystemARGS

resfinderfg v2.0:通过功能宏基因组学获得的抗生素抗性基因数据库
宏基因组学可用于监测抗生素耐药基因的扩散(ARGS)。args在诸如分解和纸牌原理等数据库中发现的源自可培养和致病性细菌,而来自不可培养和非病原细菌的ARG仍然研究了。功能元素基于表型基因的选择,并且可以从具有与已知ARGS共享的潜在低认同性的不可培养的Bacteria中识别出ARG。在2016年,创建了ResfinderFG V1.0数据库,以从功能性研究中收集ARG。在这里,我们介绍了数据库Resfinderfg v2.0的第二个范围,该v2.0可在基因组流行语Web服务器中心(https://cge.food.dtu.dtu.dk/ services/ resfinderfg/)中获得。它包括3913 ARG,由50个精心策划的数据集的功能性宏基因组学鉴定。我们评估了与肠道,土壤和水(海洋 +淡水)全球微型基因目录(https://gmgc.embl.de)相比,我们评估了其检测ARG的潜力。res- finderfg v2.0允许检测未检测到使用其他数据库检测的ARG。这些包括对β-甲酰胺,环素,苯酚,糖肽 /环烯烯和甲氧苄啶 /磺胺酰胺的抗性。因此,ResfinderFG v2.0可用于识别与常规数据库中发现的ARG,从而改善了抗抗性的描述。

珠江上游万峰湖抗生素耐药基因特征及微生物群落分布
本研究以中国科学院上海海洋大学环境科学与工程学院为研究背景,采用固相萃取、实时荧光定量PCR和宏基因组学方法进行分析。本研究的主要内容为:(1)探讨万峰湖抗生素及耐药基因的赋存特征;(2)了解沉积物中微生物群落的结构组成;(3)分析环境因素及抗生素对耐药基因分布的影响,探究抗生素及耐药基因与沉积物中微生物的共现关系。本研究结果揭示了珠江上游万峰湖抗生素及耐药基因的分布特征及沉积物微生物群落,探讨了抗生素、耐药基因与微生物之间的关系。

通过沉积DNA分析亚热带富营养湖
研究沉积档案中抗生素耐药基因(ARGS)的发生提供了重建历史(即非人性化来源)Args的分布和传播的机会。尽管在淡水环境中的ARG引起了极大的关注,但几个世纪以来几个世纪以来,多样性和丰富性的历史差异仍然很大程度上是未知的。在这项研究中,我们研究了过去600年的成谷湖沉积物中发现的细菌群落,ARG和移动遗传因素(MGE)的垂直变化模式。在保存在沉积物中的抵抗中,发现177个Args亚型,氨基糖苷和多药耐药性最丰富。上层沉积物层中的Arg丰度(等效于1940年代以来抗生素时代)低于抗生素时代期间的Arg丰度,而在后抗生体时代,ARG的多样性较高,可能是因为在最近的几十年中,人类诱导的综合疗法促进了BAC的促进和替代品的剂量。统计分析表明,MGE的丰度和细菌群落结构与ARG的丰度和多样性显着相关,这表明ARG的发生和分散性可能会通过MGE在不同细菌之间传递。我们的结果为淡水环境中ARG的自然历史提供了新的观点,对于理解暂时性的基因和ARG的传播至关重要。

冰川恢复后,天然原发性植被中的天然原发性植被
在过去的几十年中,抗生素耐药基因的传播对人类健康构成了重大威胁。尽管植物层代表了至关重要的微生物库,但对人类干扰较少的自然栖息地中ARG的概况和驱动因素知之甚少。为了最大程度地减少环境因素的影响,我们在这里收集了从初级植被继承序列的早期,中和晚期阶段收集的叶片样品,以研究植物层在自然栖息地中如何发展。拟层gr。细菌 - 养分和叶片营养素含量,以评估其对植物圈args的贡献。总共确定了151个独特的ARG,涵盖了几乎所有公认的主要抗生素类别。我们进一步发现,由于植物圈的波动栖息地和植物个体的特定选择效应,在植物群落继承过程中存在一些随机和核心集。由于植物群落继承过程中植物层细菌的多样性,综合性的复杂性和叶片养分含量的减少,Arg的丰度大大减少。虽然土壤和落叶之间的紧密联系导致叶子中的arg丰度比新鲜的叶子更高。总而言之,我们的研究表明,植物圈在自然环境中拥有广泛的ARG。这些植物层args由各种环境因素驱动,包括植物群落组成,宿主叶特性和植物圈微生物组。
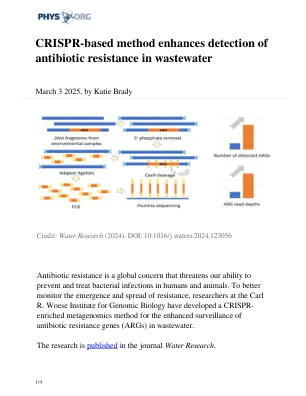
基于CRISPR的方法增强了废水中抗生素耐药性的检测
废水包含许多不同的ARG与来自人类,病毒和细菌在内的各种来源的遗传物质混合在一起。因为ARG仅占总DNA含量的很小比例,因此在废水样品中发现它们需要敏感的检测方法。最常见的技术是定量聚合酶链反应(QPCR)。此方法使用称为引物的RNA指南来识别已知ARG的特定DNA序列,然后将其放大以进行检测。

对表面沉积物中微生物群落的宏基因组学和培养基分析,以及原始河中抗生素抗性基因的流行:
摘要:居住在河流地区沉积环境中的微生物群落是原始河流生态系统的关键指标。虽然已经建立了抗生素抗性与致病性与核心肠道细菌之间的相关性,但存在着一个很大的知识差距,即抗生素抗性基因(ARGS)与人类病原细菌(HPB)与河流中的特定微生物的相互作用,通常引用了“ terrestrial terestrial gut”。在自然栖息地内,了解微生物组成,包括细菌和居民遗传因素,例如ARGS,HPB,移动遗传因素(MGE)和毒力因子(VFS)(VFS),在全球变化的背景下是必须的。为了解决这一差距,在本研究中进行了一种基于富集的培养基互补培养物和宏基因组学,以表征微生物生物库,并提供初步的生态见解,以介绍兰坎河源流域中ARG的传播。根据我们的发现,在兰开河源盆地的主流中,有674种细菌菌株在厌氧条件下包括540个菌株,在有氧条件下有124个菌株,已成功地分离出来。其中,有98种被确定为已知物种,而4种是潜在的新物种。在这98种中,有30种与人类健康有关的HPB。此外,Baca和Bacitracin分别作为该河中最丰富的ARG和抗生素出现。此外,对ARGS的风险评估主要表明危害人类健康的风险等级(等级IV)。总而言之,基于富集的培养基学被证明可有效分离稀有和未知细菌,尤其是在厌氧条件下。ARG的出现显示与MGE的相关性有限,表明对兰开河源源盆地主流内人类健康的威胁很小。

balrog-mon:用于基因组牛津纳米孔
宏基因组测序是一种最近可行的方法,可以同时表征样品中的ARG,微生物组和病原体的数据,与分离和培养细菌相比,它是一种更有效,更全面的方法。对宏基因组数据的典型分析涉及一种基于组装的方法或基于读取的方法,每种方法都有其自身的好处和限制。宏基因组装配允许对ARGS进行上游或下游研究,并提供对其起源的准确识别。但是,这种方法可能导致信息丢失,因为低覆盖的基因组通常不会组装。相比之下,基于读取的方法可实现所有可用数据的映射,但缺乏探索周围基因组环境或提供准确分类分类的能力。为了应对这些挑战,我们开发了Balrog-mon,这是一种多功能且可重现的NextFlow管道,用于测量病原体和元基因组长阅读测序的ARG,提供“组装”和“无装配”工作流程选项。

对基于 AI 的质粒注释工具进行基准测试,以便从哥斯达黎加维里拉河的宏基因组中挖掘抗生素抗性基因
摘要 — 生物信息学和人工智能 (AI) 是快速发展的工具,它们促进了移动遗传元素 (MGE) 的注释,从而能够预测污染环境中的健康风险因素,例如抗生素抗性基因 (ARG)。本研究旨在评估四种基于 AI 的质粒注释工具 (Plasflow、Platon、RFPlasmid 和 PlasForest) 的性能,通过使用定义的性能参数来识别从哥斯达黎加维里拉河获得的一个沉积物样本的宏基因组中的 ARG。我们从样本中提取并测序完整的 DNA,组装宏基因组,然后使用每种生物信息学工具进行质粒预测,并使用抗性基因标识符网络门户进行 ARG 注释。计算了评估质粒的每个 ARG 预测结果的灵敏度、特异性、精确度、阴性预测值、准确度和 F1 分数。值得注意的是,Platon 在评估的工具中表现最高,获得了优异的分数。相反,Plasflow 似乎难以区分染色体和质粒序列,而 PlasForest 在处理小重叠群时遇到了限制。RF- Plasmid 表现出较低的特异性,并且其分类单元依赖的工作流程表现不佳。我们建议采用 Platon 作为抗性基因组研究的首选生物信息学工具


对长期施用生物固体的土壤中丛枝菌根真菌接种效果的新见解:强调抗生素和
施用生物固体可以提高土壤肥力和作物产量,但也伴随着重金属和抗生素引入的风险。在重金属污染环境下,利用丛枝菌根真菌 (AMF) 是一种有效的策略,可以增强土壤微生物群落稳定性和植物对重金属的耐受性,并减少抗生素抗性基因 (ARG) 的传播。本研究通过盆栽试验探究了接种 AMF 对土壤和植物重金属含量以及土壤微生物群落的影响。结果表明,接种 AMF 显著提高了植物生物量,并降低了土壤和植物重金属含量。虽然接种 AMF 不会改变细菌和真菌群落的组成,但在较高的生物固体浓度下,它增加了细菌的多样性。值得注意的是,接种 AMF 增强了微生物网络的复杂性,并增加了关键类群的丰度。此外,在接种 AMF 的土壤中,一些对重金属具有高抗性的有益微生物得到了富集。宏基因组分析显示,与未接种AMF的土壤相比,接种AMF的土壤中移动遗传元件(MGE)基因IS91减少,重金属抗性基因增加。MGE介导的耐药基因(ARG)扩散减少的可能性是本研究的主要发现之一。需要注意的是,本研究还检测到接种AMF的高生物固体改良土壤中少数耐药基因的富集。总体而言,接种AMF可能是一种有效的农业策略,可以减轻与生物固体、重金属和抗生素耐药性相关的环境风险,从而促进可持续的土壤管理和健康。