XiaoMi-AI文件搜索系统
World File Search SystemAnion

用于阴离子交换膜燃料电池的氟化聚(芳基哌啶)膜
阴离子交换膜燃料电池 (AEMFC) 是质子交换膜燃料电池 (PEMFC) 的一种经济高效的替代品。高性能耐用的 AEMFC 的开发需要高导电性和坚固的阴离子交换膜 (AEM)。然而,AEM 通常在导电性和尺寸稳定性之间表现出权衡。本文报道了一种氟化策略,用于在聚(芳基哌啶)AEM 中创建相分离的形态结构。高度疏水的全氟烷基侧链增强了相分离,从而构建了用于阴离子传输的互连亲水通道。因此,这些氟化 PAP (FPAP) AEM 同时具有高电导率(80°C 时 > 150 mS cm − 1)和高尺寸稳定性(80°C 时溶胀率 < 20%)、优异的机械性能(拉伸强度 > 80 MPa 和断裂伸长率 > 40%)和化学稳定性(80°C 时在 3 m KOH 中 > 2000 小时)。使用本 FPAP AEM 的具有非贵重 Co-Mn 尖晶石阴极的 AEMFC 实现了 1.31 W cm − 2 的出色峰值功率密度。在 0.2 A cm − 2 的恒定电流密度下,AEM 在燃料电池运行 500 小时后保持稳定。

阴离子交换膜水电解用不锈钢316L阳极的活化
对可再生能源的日益重视导致氢和电池研究的研发工作激增。阳极析氧反应 (OER) 周围的密集电化学环境困扰着催化层、基底和多孔传输层的活性和稳定性,最终影响这两个行业。在此,我们报告了电位循环 (PC) 316L 不锈钢毡多孔传输层 (PTL) 用于阴离子交换膜水电解的好处。如 SEM、EDS、XPS、XRD 和拉曼光谱所示,PC 增加了表面粗糙度并通过铁的氧化产生了 CrFe 5 Ni 2 -O x H y 层。在三电极设置中进行的 PC 后测试显示极化电阻下降了约 68%,这反映在其用作阴离子交换膜水电解器 (AEMWE) 中的阳极时的性能上。总体而言,在阳极条件下对 PTL 进行电位循环在 AEMWE 中测试时可提高性能。可以考虑对不锈钢阳极实施这种处理,以提高 AEMWE 性能。

巨噬细胞中对OSMO敏感的LRRC8阴离子通道的激活对于微晶蛋白关节炎症很重要
沉积单钠和焦磷酸钙(MSU和CPP)微晶体负责痛风和软骨钙化中的疼痛和复发性炎症。在这些病理学中,炎症反应是由于巨噬细胞的激活引起的,负责释放包括IL-1β在内的各种细胞因子。IL-1β的成熟是由多蛋白质NLRP3插度介导的。在这里,我们发现晶体通过晶体的激活和IL-1β的同时产生的激活取决于细胞体积通过激活OSMO敏感的LRRC8阴离子通道的调节。LRC8的药理抑制和遗传沉默消除了晶体在体外和晶体诱导的胞内肿块模型中的浮游性激活。MSU/CPP晶体暴露时LRRC8激活诱导ATP释放,P2Y受体的激活和NLRP3炎性流向膜体激活和IL-1β成熟所必需的细胞内钙升高。在关节晶体诱导的炎症的背景下,我们确定了LRRC8 OSMO敏感的阴离子Channels具有病理生理相关性的功能。

肠上皮阴离子交换剂SLC26A3(DRA)的小分子抑制剂,具有腔内,细胞外作用部位
在结肠中肠上皮细胞的腔膜中表达了阴离子交换器蛋白SLC26A3(在腺瘤中下调),在那里它促进了Cl-和草酸盐的吸收。我们先前鉴定出从SLC26A3细胞质表面起作用的SLC26A3抑制剂的4,8-二甲基氨基菜蛋白类,并在小鼠的便秘模型和高氧化尿症模型中证明了它们的功效。在此,对主要筛选的50,000种新化合物和1740种活性化合物的化学类似物筛选产生了五种新型的SLC26A3选择性抑制剂(1,3-二氧二氨基氨基氨基酰胺; n- n-; n-(5-磺胺1,3,3,4- thiAdiAdiAdiAzol-2- yl-yl-yl-yl-yl-yl-yl-yl-yl-pir); 3-羧基-2-苯基苯并呋喃和苯唑嗪-4-一个),IC 50降至100 nm。动力学冲洗和作用研究发作揭示了噻唑洛 - 吡啶二肽-5-one和3-羧基-2-苯基苯甲酰苯甲氟烷抑制剂的细胞外作用部位。分子对接计算显示这些抑制剂的假定结合位点。在小鼠的洛陶化胺模型中,口服的7-(2-氯 - 苯甲基甲基)-3-苯基噻唑洛洛[3,2-A]吡啶蛋白-5-酮(3A)显着增加了粪便的体重,颗粒的数量和水含量。SLC26A3具有细胞外部作用部位的抑制剂提供了可能在口服后产生最小的全身性暴露的非吸收性,发光作用抑制剂的可能性。我们的发现还表明,可以鉴定出具有细胞外作用部位的相关SLC26阴离子转运蛋白的抑制剂,以用于对选定上皮离子运输过程的药理调节。

NaxNiyMn1-yO2 阴极中氧阴离子氧化还原活性与平面蜂窝状阳离子排序的相关性
此外,当 TMO 充电至更高电压时,晶格氧可以参与阴离子氧化还原以补偿电荷。[15,16] 因此,氧化还原反应会在首次充电时贡献额外的容量。由于晶格结构内的氧损失,相关容量在接下来的循环中通常可逆性要低得多。[17-19] 此外,过渡金属离子可以在晶格氧氧化还原反应过程中迁移到钠离子层,导致层状 TMO 的结构变形。[20,21] 因此,高能量密度 SIB 正极设计需要了解层状 TMO 中的氧阴离子氧化还原活性,以更好地设计正极材料,提高氧化还原活性的可逆性,从而稳定循环性能。层状钠 TMO 的晶格氧氧化还原活性已通过多种原位或非原位技术进行了表征,例如拉曼光谱、X 射线光电子光谱和 X 射线吸收光谱。[22 – 24] 结果通常揭示有关充电或放电时表面氧局部电子态变化的信息。[18,25,26] 此外,了解本体(晶格)氧氧化还原活性对于解释相关的晶格结构变化和电化学过程的可逆性至关重要。

大豆蛋白分离株的基于氨基酸金属络合物,用于清除超氧化阴离子自由基
抽象的超氧阴离子(O 2• - )是有害的活性氧(ROS)。跨性金属离子复合物通常被用作消除ROS的抗氧化剂。在这项工作中,首先通过氢键与聚乙烯基醇结合了大豆蛋白分离株(SPI),是一种可生物降解的蔬菜蛋白,以合成基于SPI的聚合物微凝胶(SPI-PMG)载体。此外,通过结合4-羟基水杨酸氨基酸Schiff-bas bas bas Metal Metal Complacees(Hosalcysm,M = Cu,Zn),制备了一种新型水溶性的生物聚合物/金属复合物(SCM@SPI-PMG)。SPI-PMG的结构,形态和稳定性的特征是傅立叶变换红外光谱,扫描电子显微镜,X射线衍射模式和热量分析。结果表明,获得的SPMG的直径范围为150至400 nm。此外,通过氮气四唑轻还原测定法确定了生物聚合物 - 金属配合物的清除超氧化阴离子自由基活性。与载体SPI-PMG相比,SCM@SPI-PMG的清除活动得到了极大的改进。值得注意的是,SCCU@SPI-PMG的超氧化物歧化酶(SOD)模拟达到297.10%,SCZN@SPI-PMG模拟达到35.13%。因此,SCCU@SPI-PMG可以被视为酶SOD的生物功能模仿,并且在抗氧化药物领域具有有希望的应用前景。

研究了来自原子分子动力学模拟的阴离子交换膜中氢氧离子传输的相关性
摘要:阴离子交换膜为更昂贵的质子交换膜燃料电池提供了有希望的替代品。但是,对阴离子交换膜中的氢氧化离子电导率知之甚少。在本文中,我们使用经典的分子动力学模拟来研究由乙烯 - 二乙烯基乙酸(EVA)制备的四种不同聚乙烯膜的结构和离子传输性能。我们检查了膜的微观结构,发现与具有广泛空腔分布的膜相比,腔尺寸分布狭窄的聚合物在氢氧化离子周围的水分子堆积更紧。我们计算水合膜的结构因子,并找到1和4 nm -1之间的峰,这是这些材料中离子簇的特征。我们估计水和氢氧化物离子的自扩散系数,发现水分子在所有系统中的扩散量高于氢氧化离子。氢氧化物扩散的趋势与实验电导率测量很好地对齐。对于具有广泛空腔的系统,水促进了通过车辆运输的氢氧化物扩散,并且在空腔狭窄的系统中,观察到离子跳和车辆运输。通过计算离子 - 离子和离子 - 溶剂相关性通过Onsager传输系数框架来量化这一点。关键字:聚合物膜,离子交换,分子动力学模拟,氢氧化物传输,离子体■简介

杂原子掺杂的碳基电催化剂的最新进展用于阴离子交换膜燃料电池中的氧气还原反应
简介。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>3594碱性培养基中还原反应。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3594 ORR在碱性培养基中的一般原理和机制。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3595个在阴离子交换膜燃料电池中的ORR的电催化剂。 。 。 。 。 。 。 。 。 。 。 。 。 。 。 。 。 。 。 。3595个在阴离子交换膜燃料电池中的ORR的电催化剂。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3598碳纳米管。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3598石墨烯。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3599生物质量衍生的碳。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3599杂种掺杂的碳设计和合成。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。 div>。 div>3599氮气cnts。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 3601硼偏用的中枢神经系统。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div> 。 div>3599氮气cnts。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>3601硼偏用的中枢神经系统。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3605磷掺杂的CNT。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3607共同掺杂的CNT。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3607金属氮掺杂的CNT。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3610氮掺杂的谷物。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>3611:泛图。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>3611磷掺杂的谷物。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3612共掺杂/多杂种掺杂石墨烯。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3612金属,杂体共掺杂石墨烯。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3615生物启发的ORR催化剂。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3617 AMFC性能和稳定性。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>3620结论和勘探的依据。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>3621竞争利益声明。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。 div>。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3623致谢。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3623参考。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。。3623

通过阴离子选择的无机固体电解质相互重点的原位工程可实现稳定的锂阳极
发现液体电池电解质有助于促进稳定的固体电解质相互作用(SEIS)减轻树突形成,这对于在下一代能量密集的电池中启用锂阳极至关重要。与传统的电解质溶剂相比,基于四氢呋喃(THF)的电解质系统已经通过鼓励阴离子的分解(而不是有机溶剂),从而产生了无机富丽石的SEIS,从而在实现高稳定性锂阳极方面取得了巨大成功。在此,通过采用各种不同的锂盐(即LIPF 6,Litfsi,Lifsi和Lidfob),可以证明电解质阴离子会调节SEI的无机组成和产生的特性。通过新的分析时间二级离子质谱法,例如对深度促值的分层聚类和使用综合产量的组成分析,从每个电解质系统产生的SEI的化学组成和形态。值得注意的是,Lidfob电解质提供了一个异常稳定的系统,可实现锂阳极,以0.5 mAh g -1的电流密度传递> 1500个循环,在对称细胞中的容量为0.5 mAh g -1。此外,LI //使用该电解质的LFP细胞表现出高速率,可逆的锂储存,提供139 mAh g(LFP)-1
MD Anderson的白血病护理
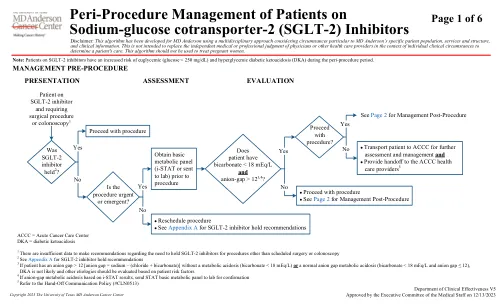
1没有足够的数据来提出有关在计划手术或结肠镜检查以外的其他程序的需求的建议。2请参阅SGLT-2抑制剂建议的附录A,如果患者的GAP gap> 12 [Anion Gap> 12 [Anion Gap = sodium + sodium + sodium + bicacoride + bicacorice + a in conbicalication/abicalication] lobiciss loseboriss/abicain dicraborice 来评估其他病因