XiaoMi-AI文件搜索系统
World File Search SystemMTBC

固定后时间的影响
摘要我们分析了迄今为止收集的结核分枝杆菌复合物(MTBC)的最多样化基因组数据集的泛基因组和基因含量调节。MTBC的封闭泛基因组的特征是辅助和特异性基因组的降低,与其克隆性质兼容。然而,随着MTBC基因组的系统发育距离的增加,共享基因家族大大少得多。这种效应仅在种间比较中观察到,而不是SPE内部的CIE,这表明物种特异性的生态特征与基因含量的变化有关。基因丢失是由于基因组缺失和伪源性元素引起的,可驱动基因含量的变异。MTBC物种和谱系之间的这种基因侵蚀也有所不同,即使在结核分枝杆菌中,L2的基因损失比L4更大。我们还表明,系统发育近端并不总是与MTBC中基因含量相关性的良好替代性,因为MyCobacte Rium Africanum L6的基因曲目偏离了其预期的系统发育生物的保守主义。以伪元注释代表的毒力因子的基因破坏大多不是保守的,是MTBC生态型的预测因素。每个MTBC生态型都具有自己的附件基因组,可能受宿主和地理等不同选择压力的影响。重要的是研究基因丧失如何赋予MTBC菌株的新适应性特征。检测到的异质基因损失在阐明负责MTBC中观察到的各种表型的遗传因素方面构成了重大挑战。通过详细介绍特定的基因损失,我们的研究是研究MTBC表型及其免疫逃避策略的研究人员的资源。

信息表:GenoScreen Deeplex Myc-TB 测试
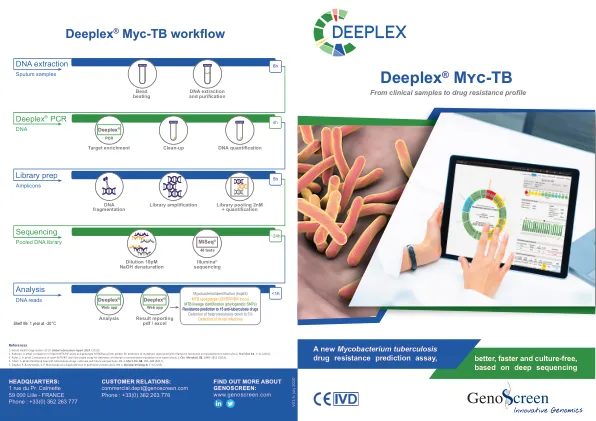
GenoScreen 拥有基于新一代测序 (NGS) 的试剂盒,可同时识别分枝杆菌种类、进行基因分型并预测结核分枝杆菌复合群 (MTBC) 菌株的耐药性;该试剂盒 (Deeplex® Myc-TB) 可直接用于临床样本 (1) 。该检测依赖于单个 24 重扩增子混合物的深度测序,针对与一线和二线抗结核药物(利福平、异烟肼、吡嗪酰胺、乙胺丁醇、氟喹诺酮类、阿米卡星、卡那霉素、卷曲霉素、链霉素、乙硫异烟胺、贝达喹啉、氯法齐明和利奈唑胺)耐药性相关的 18 个主要 MTBC 基因区域。 hsp65 基因是分枝杆菌种属识别的靶标,而 spoligotyping 靶标(CRISPR/直接重复 [DR] 基因座)和耐药相关靶标中的系统发育单核苷酸多态性 (SNP) 用于 MTBC 菌株基因分型。

范围长尾猕猴(Macaca fascicularis)
在自由行动或野生动物中对传染病的监测已在COVID-19发作之后在许多栖息地国家进行了广泛进行。泰国位于长尾猕猴(Macaca fascicularis; MF)的分布范围的中心,其中动物既经常人类接触,又有人类结核病的高患病率。用于大规模检测结核分枝杆菌复合物(MTBC)的使用为6110-MF中的pcr,使用口服(通过绳索诱饵)和粪便(直接擦拭新鲜粪便)收集标本。首先,MTBC-IS 6110被限制的PCR在非侵入性收集的标本中得到了验证,其特异性和陈述性,然后与24个圈养的MTBC诱发的MTBC诱导的MF中的口腔和直肠拭子相比。验证后,将这些方法应用于在先前报道的MTBC感染人群中的四个棚屋MTBC(MTBC)患病率的调查。总共收集了173个诱饵绳标本和204个新鲜排定的排泄物。IS 6110 -PCR技术的检测极限为10 fg/μL,181 bp PCR扩增子与MTB H37RV基因组序列显示100%序列相似性。在被俘虏的可疑MF中的侵入性和非侵入性收集的标本之间检测揭示了两种类型的口服样本之间存在显着相关性(口腔拭子和诱饵的绳索; n = 24,r 2 = 1,r 2 = 1,p-value <0.001),但较高的新鲜伪造群体比MTCRES shand Swabs shews swabs。揭示了两种类型的口服样本之间存在显着相关性(口腔拭子和诱饵的绳索; n = 24,r 2 = 1,r 2 = 1,p-value <0.001),但较高的新鲜伪造群体比MTCRES shand Swabs shews swabs。此外,在新鲜的粪便中,MTBCS阳性自由放大的MF的比例明显高于诱饵绳(5.20%; 95%CI; 95%; 95%; 95%; 4.9-12.7%)的比例。该结果表明,通过诱饵绳索和粪便采样通过排除排泄物拭子可以用作自由态非人类灵长类动物中MTBCS检测的辅助标本。

马里抗酸阳性疑似结核病患者中非结核分枝杆菌的流行情况
分枝杆菌属包括导致人类和动物结核病 (TB) 的结核分枝杆菌复合群 (MTBC) 的种、导致麻风病的麻风分枝杆菌,以及通常称为非典型或非结核分枝杆菌 (NTM) 的分枝杆菌种,其中包括导致布鲁里溃疡的溃疡分枝杆菌。与 MTBC 组成员不同,NTM 不是人类的专性寄生虫,而是土壤和水的正常居民,可以在天然水源和处理过的水源中找到 [1]。已正式确认的 NTM 有 200 多种 [2],其中已知约 25 种与人类疾病密切相关。一些种与引起类似 TB 症状的肺部疾病有关 [1]。由于它们的栖息地,人类每天都会接触到这些细菌。因此,必须将 NTM 病与简单的定植或临床样本污染(例如自来水)区分开来 [1,3]。与结核病不同,NTM 引起的疾病的全球流行病学尚不明确。从临床标本中分离 NTM 的病例主要见于工业化国家,患病率和发病率各不相同。基于肺部标本分离株的研究报告称,2004 年至 2006 年美国的患病率为每 100,000 人 1.4 至 6.6 人 [ 4 ],2010 年加拿大安大略省的患病率为每 100,000 人 9.8 人 [ 5 ],2020 年德国的患病率为每 100,000 人 5.8 人 [ 6 ]。也有报告称,2012 年英格兰的发病率为每 100,000 人 6.1 人 [ 7 ],2020 年德国的发病率为每 100,000 人 5.3 人 [ 6 ]。在结核病流行国家,NTM 的报告频率较低,并且主要发生在高危人群中,特别是具有易感条件或免疫力低下的人群 [ 8 ]。然而,工业化国家的经验表明,结核病负担的下降也增加了发现的 NTM 病例数。随着另一种环境下结核病防治规划的加强,我们或许也会看到类似的情况,对中低收入国家而言,诊断和临床治疗的挑战将日益加大[9]。NTM 肺病的诊断基于临床、放射学和微生物学标准[1]。在大多数资源有限的国家,基本上无法进行以实验室为基础的 NTM 检测,无法与 MTBC 相区分并确定其菌种。显微镜检查是最容易获得的技术,它将 MTBC 和 NTM 识别为抗酸杆菌 (AFB),但无法区分它们。自 2010 年以来,世界卫生组织 (WHO) 已推荐使用 GeneXpert MTB/RIF(Xpert)等快速分子检测作为结核病诊断的初始检测,该检测具有更高的灵敏度和特异性 [10]。该检测仅可识别样本中是否存在 MTBC 菌种。如果 AFB 阳性痰液样本经 Xpert 检测呈 MTBC 阴性,则可能提示感染 NTM [11]。在马里,已报道过 NTM 感染病例,特别是在抗结核治疗失败或结核病治愈后复发的患者中 [ 12 ]。在该国引入 Xpert 后,AFB 涂片阳性而 Xpert 检测阴性的疑似 NTM 感染病例报告更频繁 [ 13 ]。

杭州圣庭医疗科技有限公司TBSeq测试
杭州盛廷医疗科技有限公司拥有一款基于靶向二代测序(NGS)的试剂盒,用于同时识别分枝杆菌种类并预测结核分枝杆菌复合群(MTBC)菌株的耐药性。该试剂盒 TBseq® 可直接应用于痰液、支气管肺泡灌洗液、胸腔积液或分枝杆菌阳性培养物等临床标本。它依赖于引物多重扩增混合物的深度测序,针对与一线和二线抗结核(抗 TB)药物(利福平、异烟肼、吡嗪酰胺、乙胺丁醇、氟喹诺酮类、阿米卡星、卡那霉素、卷曲霉素、链霉素、对氨基水杨酸、环丝氨酸、乙硫异烟胺/丙硫异烟胺、贝达喹啉、氯法齐明和利奈唑胺)耐药相关的 21 种主要 MTBC 基因。分枝杆菌种属鉴定是通过针对 16S 和 hsp65 基因区域进行的。

Deeplex Myc-TB 技术说明
Deeplex® Myc-TB 检测依赖于与对一线和二线药物耐药性相关的 18 个主要 MTBC 基因靶点的深度测序(图 3)。根据在这些基因座中观察到的突变的存在或不存在以及对参考数据库*****的查询,预测样本中的 MTBC 菌株对每种抗生素敏感或耐药,或具有尚未表征的突变(图 1)。可以轻松查看单个靶点位置和突变及其序列覆盖深度。可通过超链接访问描述突变与耐药性关联的参考文献信息。总体而言,该检测可以预测对 15 种抗结核药物/药物类别的耐药性,包括最近推出的化合物,如贝达喹啉和利奈唑胺,使其成为迄今为止最详尽的可直接应用于样本的基因型检测。
Deeplex® Myc-TB
Deeplex® Myc-TB 检测依赖于对 18 个主要 MTBC 基因靶点进行深度测序,这些靶点与对一线和二线药物的耐药性有关(图 3)。根据在这些基因座中观察到的突变的存在或不存在情况以及对参考数据库的查询,预测样本中的 MTBC 菌株对每种抗生素敏感或耐药,或具有尚未表征的突变(图 1)。可以轻松看到各个靶点位置和突变以及它们的序列覆盖深度。可通过超链接访问描述突变与耐药性关联的参考文献信息。总的来说,该检测可以预测对 15 种抗结核药物的耐药性,包括最近推出的贝达喹啉和利奈唑胺 4 等化合物,使其成为迄今为止最详尽的可直接应用于样本的基因型检测。

驱动因素和多样性的DNA腺嘌呤甲基甲基甲基甲基甲基甲基甲基甲基甲基甲基甲基甲基甲基甲基甲基酸酯复合物临床分离株
摘要这项研究组装了93种结核分枝杆菌复合物(MTBC)分离株的DNA腺嘌呤甲基组,并从7个谱系中与完全注销的,完成的,从头组装的基因组配对。综合分析产生了四个关键结果。首先,甲基转移酶 - 甲基甲基映射校正的甲基转移酶变体效应以前被基于参考的变体调用遮盖。第二,部分活性的甲基转移酶等位基因的异质性分析表明,细胞内随机甲基化在等生培养物中产生甲基甲基化合物的镶嵌性,我们将其形式化为“细胞间摩西甲基化”(IMM)。突变驱动的IMM在全球突出的北京sublineage中几乎无处不在。第三,启动子甲基化是广泛的,与D HSDM转录组中的差异表达相关,表明启动子HSDM甲基化直接影响转录。最后,比较和功能分析确定了351个位点可在分离株和许多推定的调节相互作用之间进行高变量。这种多摩变整合揭示了临床分离株中甲基甲基变异性的特征,并为假设DNA腺嘌呤甲基化在MTBC生理学和适应性进化中的功能提供了合理的基础。

Deeplex® Myc-TB - 用户手册
该检测依赖于与一线和二线药物耐药性相关的 18 种主要 MTBC 基因靶标的深度测序(表 2)。可以在高读取深度下对分枝杆菌基因靶标进行测序。因此,每个序列位置都可以被许多读取覆盖,从而实现高度可信的突变调用。可以在占样本中细菌 3%(在位置特定读取深度条件下甚至为 1%)的突变/异质耐药亚群中检测到变异,这是其他快速分子检测无法实现的。可以表征代表低至 100-1000 个分枝杆菌基因组/杆菌的提取 DNA,因此低于传统显微镜的检测限。该试剂盒还包括对安全 Web 应用程序 Deeplex® Web 应用程序的访问,以便快速轻松地分析和解释测序数据。

结核分枝杆菌复合药物敏感性测试中的问题:乙胺丁醇
EMB 抗性菌株的最低抑菌浓度 (MIC) 往往在 7.5 μg/mL 至 40 μg/mL 范围内。8–11 5 μg/mL 的测试浓度(使用分枝杆菌生长指示管 (MGIT))可以区分大多数敏感菌株和抗性菌株。除了传统的基于生长的药物敏感性测试 (DST) 之外,DNA 突变的分子检测也可以提供预测耐药性的宝贵信息。虽然 MTBC 对 EMB 的耐药机制尚不明确,基因组靶点也未得到充分记录,12 但许多研究人员已将研究重点放在 embCAB 操纵子的作用上,特别是 embB 基因。多名研究人员发现,embB 密码子 306 的突变是最常见的点突变,50–70% 的分离株含有赋予 EMB 抗性的突变。 5,8,11,13–16 然而,embB 中的其他突变,以及 embC 和 embA 中的突变,也已被证实