XiaoMi-AI文件搜索系统
World File Search SystemNPC1

尼曼 - pick型疾病中的内聚糖功能障碍和神经元 - 斑点串扰
niemann - pick型(NPC)疾病是一种罕见的进行性溶酶体脂质储存障碍,表现出具有临床综合症的异质谱,包括内脏,神经系统和精神症状。这种单基因常染色体隐性疾病主要是由控制细胞内脂质稳态的NPC1基因中的突变引起的。囊泡介导的内糖体脂质运输和通过轨道间膜接触位点通过孔间膜接触位点的非西西脂质交换。NPC1功能的丧失会触发各种脂质物种的细胞内积累,包括胆固醇,糖磷脂,鞘磷脂和鞘氨醇。NPC1介导的脂质转运功能障碍对所有脑细胞都有严重的后果,从而导致神经变性。除了神经元NPC1的细胞自主贡献外,其他脑细胞中异常的NPC1信号对于病理至关重要。我们在这里讨论NPC病理学中神经元,少突胶质细胞,星形胶质细胞和小胶质细胞之间的内染色体功能障碍和Atight串扰的重要性。我们坚信,特定细胞的救援可能不足以抵消NPC病理的严重程度,而是针对常见机制(例如内部溶酶体和脂质运输功能障碍)可能会改善NPC病理学。本文是讨论会议问题的一部分,“理解神经变性中的内聚糖网络”。


HMS-SNUH-SNUCM利益信Salic
受影响的产物积聚在溶酶体中,导致溶酶体功能障碍和随之而来的疾病。NPC1和NPC2是溶酶体途径的一部分,用于疏散胆固醇,该胆固醇因内吞作用所吸收的脂蛋白分解而产生(图1A)。如果该途径受到NPC1或NPC2突变的损害,则胆固醇会积聚在溶酶体中,从而导致NPC。目前尚无对NPC的有效治疗方法。我基于绕过NPC1/2途径的溶酶体靶向融合的简单但新颖的NPC治疗方法(图1B)。简要地,NPC2易于表达和净化,并在添加到细胞中时有效地靶向溶酶体。通过将NPC2结合到无毒的胆固醇结合循环寡糖(例如β-环糊精(B CD))中,我们将产生一种融合,与单独的NPC2不同,可以将胆固醇直接捐赠给溶酶体膜,从而校正Lysosomal sostemos,从而纠正Lysosomal sostemot(Fig.1b)。有两年的资金,我建议实现以下目的:1)生成一组NPC2-B CD共轭物,其B CD部分变化以及NPC2和B CD之间的连接链接器; 2)确认靶向NPC2-B CD偶联物与溶酶体的靶向; 3)测试NPC2-B CD偶联物对NPC患者成纤维细胞中溶酶体胆固醇积累的影响,以确定它们是否以及如何促进胆固醇流动性。

多路复用DNA-Paint成像晚期/溶酶体蛋白组成的异质性
晚期内体/溶酶体(LELS)对于许多生理过程至关重要,它们的功能障碍与许多疾病有关。蛋白质组学分析已经鉴定出数百种LEL蛋白,但是,这些蛋白是否均匀地存在于每个LEL上,或者是否存在具有独特蛋白质组成的细胞类型依赖性LEL亚群,尚不清楚。我们采用了定量的多重DNA-油漆超分辨率方法来检查单个LELS上六种关键LEL蛋白(Lamp1,Lamp2,CD63,TMEM192,NPC1和LAMTOR4)的分布。虽然LAMP1和LAMP2在LEL中含量丰富,但标志着公共种群,大多数分析的蛋白质与特定的LEL亚群有关。我们的多重成像方法基于其独特的膜蛋白组成,最多鉴定出多达八个不同的LEL亚群。此外,我们对这些亚群和线粒体之间的空间关系的分析表明,NPC1阳性LELS的细胞类型特异性趋势与线粒体紧密地位。我们的方法将广泛适用于在许多生物学环境中用单细胞器分辨率来确定细胞器异质性。
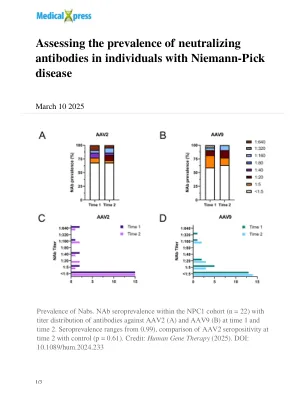
评估Niemann-Pick疾病个体中和抗体的患病率
“这样的研究对于一般和NPC1患者的基因疗法治疗的分娩策略非常重要,例如确定试验的入学标准,治疗方案是否需要减轻先前存在的NAB,以及在生活过程中NAB的发展可能会影响Hull of Human Gene edior of Munly Gention of Munly Gention of Hull of Munly Gention of Hull of Munly Gention of Hull of Hull of Hull of Hull of Hull of Human Gention for Human Gention of Hull of Hull of Hull of Hull of Hull of Hull of Hull of Hull of Hull of the the。马萨诸塞州chan

使用CRISPR Prime编辑
使用CRISPR Prime编辑Steven Erwood 1,2,Teija M.I.的饱和变体解释。bily 2,†,Jason Lequyer 1,3,†,Joyce Yan 2,Nitya Gulati 1,2,Reid A.Brewer 2,4,Liangchi Zhou 2,Laurence Pelletier 1,3,Evgueni A. Ivakine 2,4,*,Ronald D. Cohn 1,2,4,5 1。加拿大多伦多多伦多大学分子遗传学系2.遗传学和基因组生物学计划,加拿大安大略省多伦多的病儿童研究所医院3.Lunenfeld-Tanenbaum研究所,加拿大安大略省多伦多山医院4.加拿大多伦多多伦多大学生理学系5。多伦多大学儿科和生病儿童医院,加拿大多伦多的医院†这些作者在过去十年中向Zhenya.ivakine@sickkids.ca摘要贡献了同样的贡献,在过去的十年中,下一代测序在临床实践中已广泛实施。 然而,由于经常确定具有不确定意义的遗传变异(VU),因此对这种变体的缩放功能解释的需求变得越来越明显。 一种解决此问题的方法是饱和基因组编辑(SGE),它允许对单核苷酸变体进行缩放的多重功能评估。 但是,SGE的当前应用依赖于同源指导的维修(HDR)引入感兴趣的变体,这受到较低的编辑效率和低产品纯度的限制。 此外,我们设计了一种基因组编辑策略,该策略允许基因基因座的单倍体化,该策略允许几乎任何细胞类型中的孤立变体解释。多伦多大学儿科和生病儿童医院,加拿大多伦多的医院†这些作者在过去十年中向Zhenya.ivakine@sickkids.ca摘要贡献了同样的贡献,在过去的十年中,下一代测序在临床实践中已广泛实施。然而,由于经常确定具有不确定意义的遗传变异(VU),因此对这种变体的缩放功能解释的需求变得越来越明显。一种解决此问题的方法是饱和基因组编辑(SGE),它允许对单核苷酸变体进行缩放的多重功能评估。但是,SGE的当前应用依赖于同源指导的维修(HDR)引入感兴趣的变体,这受到较低的编辑效率和低产品纯度的限制。此外,我们设计了一种基因组编辑策略,该策略允许基因基因座的单倍体化,该策略允许几乎任何细胞类型中的孤立变体解释。在这里,我们对SGE进行了改编的CRISPR质量编辑,并证明了其在理解溶酶体储存障碍尼曼 - 佩克病C1(NPC)的NPC1基因中变体的功能意义的实用性。通过将饱和素编辑(SPE)与临床相关的测定相结合,我们在NPC1单倍体HEK293T细胞中的功能评分和解释了256种变体。为了进一步证明该策略的适用性,我们使用SPE和细胞模型单倍体化在BRCA2基因中的功能上为465个变体分数。我们预计我们的工作将可以翻译成具有适当的细胞测定法的任何基因,从而可以更快,准确地诊断,改善遗传咨询,并最终确切地精确的患者护理。引言精度或个性化药物必然是基于对人群中发现的遗传变异的强烈理解。因此,人类疾病基因中发现的VU的优势是实现

Niemann – Pick疾病中的遗传基础,肺参与和治疗选择:全面的评论
摘要:Niemann – Pick疾病(NPD)是属于溶酶体储存障碍的罕见常染色体隐性疾病。已经描述了三种类型的NPD:NPD A型,B和C型A型A和B型是由编码鞘磷脂磷酸二酯酶1的基因SMPD1中的突变引起的,因此缺乏酸性鞘磷脂酶活性。这些疾病已被归类为酸鞘磷脂酶缺陷(ASMDS)。NPD C型是由于基因NPC1或NPC2的突变而导致的一种神经系统疾病,导致胆固醇运输和酯化的缺陷。尽管所有三种NPD都可以表现出肺部受累,但肺部疾病在NPD B型中更频繁地发生,通常患有间质肺部疾病,复发性肺部感染和呼吸衰竭。从这个意义上讲,带有支气管 - 肺泡灌洗或活检以及高分辨率计算机断层扫描的支气管镜检查是基本的诊断工具。迄今为止,已经做出了一些努力,为NPD找到有效的疗法,但只有有限的治疗选择。用olipudaseα的酶替代疗法是ASMD患者的第一个也是唯一批准的疾病改良疗法。文献中的ASMD还描述了肺移植和造血干细胞移植。NPD C型中唯一认可的疾病改良疗法是Miglustat,一种底物还原治疗。这篇综述的目的是在遗传基础和肺参与NPD的基础上描述一种最新的现状,重点关注疾病的临床表现,放射学和组织病理学特征,以及可用的治疗选择,并注视着未来治疗策略。