XiaoMi-AI文件搜索系统
World File Search SystemSmiles
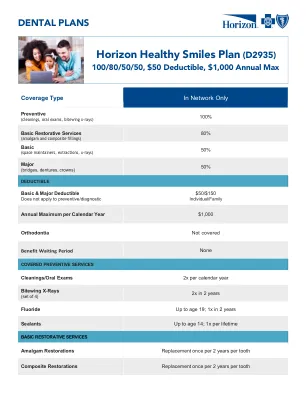
地平线健康微笑计划 (D2935)
本文件仅供参考,不构成具有约束力的协议。请注意,费率可能会发生变化。请联系 Horizon 获取最新费率。本文件提供的信息并非旨在取代或修改 Horizon 颁发或管理的健康、牙科或视力福利计划中包含的条款、条件、限制和排除条款。如果本文件中包含的信息与您的计划文件发生冲突,则以您的计划文件为准。新泽西州 Horizon Blue Cross Blue Shield 是蓝十字蓝盾协会的独立被许可人。Blue Cross ® 和 Blue Shield ® 名称和符号是蓝十字蓝盾协会的注册商标。Horizon ® 名称和符号是新泽西州 Horizon Blue Cross Blue Shield 的注册商标。© 2021 新泽西州 Horizon Blue Cross Blue Shield,Three Penn Plaza East,新泽西州纽瓦克 07105。| Horizon Blue Cross Blue Shield of New J ersey 遵守适用的联邦民权法律,不会因种族、肤色、性别、国籍、年龄、残疾、怀孕、性别认同、性别、性取向或健康状况在计划管理(包括登记和福利决定)中歧视、排斥或区别对待任何人。西班牙语 (Español): 如需西班牙语帮助,请拨打 1-866-660-6528 (TTY 711 )。中文 (中文): 如需中文帮助,请致电 1-866-660-6528 (TTY 711 )。ECNA005423 (1221)

健康的款项安大略省牙科服务和费用的时间表
www.ontario.ca/healthysmiles(英语)或www.ontario.ca/beauxsouririres(法语)或从其当地的公共卫生部门或安大略省的地方获得申请表。完成的申请必须邮寄至:

使用反应微笑对原子经济进行定量分析:一种计算方法†/div>
我们提出了一种用于使用反应微笑来计算化学反应的原子经济算法的实施。Python编程用于连接RDKIT库来解析和解释化学结构,从而提供准确有效的化学可持续性计算。通过实施强大的算法来处理化学计量系数和多种反应,该方法对原子经济进行了全面的分析,这是绿色化学实践必不可少的指标。此外,这种计算方法可以轻松地集成到产生大量化学反应的AI应用中,作为筛选和优化步骤,进一步增强了可持续化学过程设计的潜力。我们通过几个案例研究证明了它的应用,强调了其有助于设计更可持续的化学过程的潜力。我们使用阿司匹林及其多个合成路线证明了这种方法。

基于SMILES的分子生成模型在新药设计中的应用
在人类与疾病的长期斗争中,药物发挥着越来越重要的作用。药物发现是识别潜在的新治疗实体的过程,而药物设计是基于对生物靶标的了解寻找新药物的过程,涉及分子的设计(Zhou and Zhong,2017)。药物发现和设计一直面临障碍,因为需要大量的人力、物力和财力。随着人工智能在图像处理、模式识别和自然语言处理等领域的成功(Xie et al.,2022),深度生成模型在药物发现领域引起了广泛关注,同时在分子设计优化领域也展现出良好的应用前景。当使用生成模型生成分子时,其实质是学习训练集中分子的分布,然后生成与训练集中分子相似但不同的分子。结合进化算法或强化学习,可以进一步优化生成分子的性质(Tong et al.,2021;Tan et al.,2022a)。生成模型中的分子表示可以有多种形式,包括简化的分子输入行输入系统(SMILES)、分子图等。生成模型大致可分为五类,包括循环神经网络(RNN)、自编码器(AE)、生成攻击网络(GAN)、Transformer和结合强化学习(RL)的生成模型(Bhisetti and Fang,2022),如图1A所示。其中基于文本序列的分子生成模型(SMILES)应用最为广泛。本文简单介绍基于最新的文本序列分子设计(SMILES)的深度生成模型的基本原理及应用,以便读者了解深度生成模型并将其更好地运用在药物分子设计中。

TarDict:基于 RandomForestClassifier 的软件预测药物-目标相互作用
根据已确定药物的数据预测未知或/和正在研究的药物的靶标不仅对于理解各种药物和分子相互作用过程非常重要,而且对于开发新药也非常重要。这里我们介绍 TarDict,一种基于 RandomForestClassifier 的软件,它基于化学物质的 SMILES 预测靶标通路或蛋白质。TarDict 接收 SMILES 并返回可能类似药物的列表,然后向用户导出药物所起作用的靶标列表。20442 个条目的训练数据集和测试显示准确率为 %95。



使用以 Morgan 指纹为特征的深度神经网络模型预测 ErbB 抑制剂的结合亲和力
ErbB 受体家族(包括 EGFR 和 HER2)在细胞生长和存活中起着至关重要的作用,并与乳腺癌和肺癌等各种癌症的进展有关。在本研究中,我们开发了一个深度学习模型,使用基于 SMILES 表示的分子指纹来预测 ErbB 抑制剂的结合亲和力。每种 ErbB 抑制剂的 SMILES 表示均来自 ChEMBL 数据库。我们首先从 SMILES 字符串生成 Morgan 指纹,并应用 AutoDock Vina 对接来计算结合亲和力值。根据结合亲和力过滤数据集后,我们训练了一个深度神经网络 (DNN) 模型来根据分子指纹预测结合亲和力值。该模型取得了显著的性能,训练集上的均方误差 (MSE) 为 0.2591,平均绝对误差 (MAE) 为 0.3658,R 平方 (R²) 值为 0.9389。尽管在测试集上性能略有下降(R² = 0.7731),但该模型仍然表现出强大的泛化能力。这些结果表明深度学习方法对于预测 ErbB 抑制剂的结合亲和力非常有效,为虚拟筛选和药物发现提供了宝贵的工具。

DeepGS:用于药物靶标结合亲和力预测的图形和序列的深度表示学习
摘要:准确预测药物-靶标结合亲和力 (DTA) 是药物发现中的一项关键任务。大多数传统的 DTA 预测方法都是基于模拟的,这严重依赖于领域知识或具有靶标的 3D 结构的假设,而这些知识通常很难获得。同时,传统的基于机器学习的方法应用各种特征和描述符,并且仅仅依赖于药物-靶标对之间的相似性。最近,随着可用的亲和力数据的增加和深度表示学习模型在各个领域的成功,深度学习技术已应用于 DTA 预测。然而,这些方法考虑了标签/独热编码或分子的拓扑结构,而没有考虑氨基酸和 SMILES 序列的局部化学背景。基于此,我们提出了一种新颖的端到端学习框架 DeepGS,该框架使用深度神经网络从氨基酸和 SMILES 序列中提取局部化学背景,以及从药物中提取分子结构。为了协助对符号数据的操作,我们建议使用先进的嵌入技术(即 Smi2Vec 和 Prot2Vec)将氨基酸和 SMILES 序列编码为分布式表示。同时,我们提出了一种在我们的框架下运行良好的新分子结构建模方法。我们进行了大量的实验,将我们提出的方法与最先进的模型(包括 KronRLS、SimBoost、DeepDTA 和 DeepCPI)进行了比较。大量的实验结果证明了 DeepGS 的优越性和竞争力。

田纳根:纯变压器编码器为从头分子的生成提供了有效的离散gan
使用离散数据(例如简化的分子输入线 - 输入系统(Smiles)字符串)的从头生成的深层生成模型吸引了药物设计中的广泛关注。然而,训练不稳定经常困扰生成的广告网络(GAN),导致可能崩溃和低偏移性等概率。这项研究提出了一个纯粹的变压器编码器GAN(宽度)来解决这些问题。宽度的发电机和鉴别剂是变压器启动器的变体,并与加固学习(RL)结合使用,以生成具有所需化学特性的分子。此外,变体微笑的数据增强是为了学习微笑字符串的范围和语法的宽度培训。在方面,我们引入了一个增强的田纳州的变体,称为十(w)gan,其中包含了微型批处理歧视,并提高了生成分子的能力。对QM9和锌数据集的实验结果和消融研究表明,所提出的模型以计算有效的方式产生了具有所需化学性质的高效和新颖的分子。