XiaoMi-AI文件搜索系统
World File Search SystemTRAES


加州大学洛杉矶分校
结果 共招募了 79 名患者;第 1a 部分招募 19 名,第 1b 部分招募 8 名,第 2 部分招募 52 名。未报告剂量限制性毒性,根据安全性、耐受性、药代动力学参数和临床活性,确定推荐剂量为每 2 周 15 mg/kg。最常见的治疗相关不良事件 (TRAE) 是疲劳 (17.7%)、恶心 (11.4%) 和干眼症 (10.1%)。3 级 TRAE 包括恶心 (2 名患者) 和贫血、中性粒细胞减少、AST 升高、碱性磷酸酶升高、呕吐和输液反应 (各 1 名患者)。在 28 名患者中,有 3 名 (10.7%) 报告了可逆性的 2 级角膜 TRAE,该组剂量为每 2 周 1 次,共 70 天。未报告 4 级以上 TRAE。28 名 FGFR2b 过表达 GEA 患者中,5 名(17.9%;95% CI,6.1% 至 36.9%)确认有部分缓解。

图 S1。OS 的森林图。这些森林图显示了不同治疗比较中 OS 的汇总风险比和 95% 可信区间。(A)
图 S5. ≥3 级 TRAE 的森林图。这些森林图显示了不同治疗比较中 ≥3 级 TRAE 发生的优势比和 95% CI。(A)PD-1/PD-L1 抑制剂与化疗的结果。(B)PD-1/PD-L1 抑制剂与安慰剂的结果。(C)PD-1/PD-L1 抑制剂联合化疗与单独化疗的结果。TRAE,治疗相关不良事件;CI,置信区间;PD-1,程序性细胞死亡-1;PD-L1,程序性死亡配体 1。

II型RAF抑制剂ToVorafenib在复发/难治性小儿低级神经胶质瘤中:2期Firefly-1试验
BRAF基因组改变是小儿低级神经胶质瘤(PLGG)中最常见的致癌驱动因素。ARM 1(n = 77)试验研究了口服,选择性,中枢神经系统 - pentrant,II型RAF抑制剂Tovorafenib(420 mg m -2一次,每周420 mg M -2最高600 mg)对BRAF -ARAF -ARETED -ARETED -ARETED -ARTERED -PLACCED的患者的功效。ARM 2(n = 60)是一个延伸队列,它为ARM 1闭合后的RAF改变PLGG患者提供了治疗。基于独立审查,根据神经肿瘤高级神经胶质瘤(RANO-HGG)标准的响应评估,67%的总响应率(ORR)符合ARM 1 1预定的主要端点;响应持续时间(DOR)为16.6个月;响应时间(TTR)的中位时间为3.0个月(次要终点)。通过小儿神经肿瘤学低级神经胶质瘤(RAPNO)标准和安全性评估评估的其他选择的次要终点包括ORR,DOR和TTR(在所有治疗的患者中评估,ARM 2,ARM 2,n = 137)。根据RAPNO标准(包括次要答复)的ORR为51%; DOR中位数为13.8个月; TTR中位数为5.3个月。最常见的治疗不良事件(TRAES)是头发颜色变化(76%),肌酸磷酸激酶(56%)和贫血(49%)。≥3级Traes发生在42%的患者中。九(7%)患者患有TRAES导致停用Tovorafenib。这些数据表明,Tovorafenib可能是对BRAF的,经过重复/难治性的PLGG的有效疗法。临床。GOV注册:NCT04775485。
初步阶段1 IMM-1-104的安全性和活性,AN ...
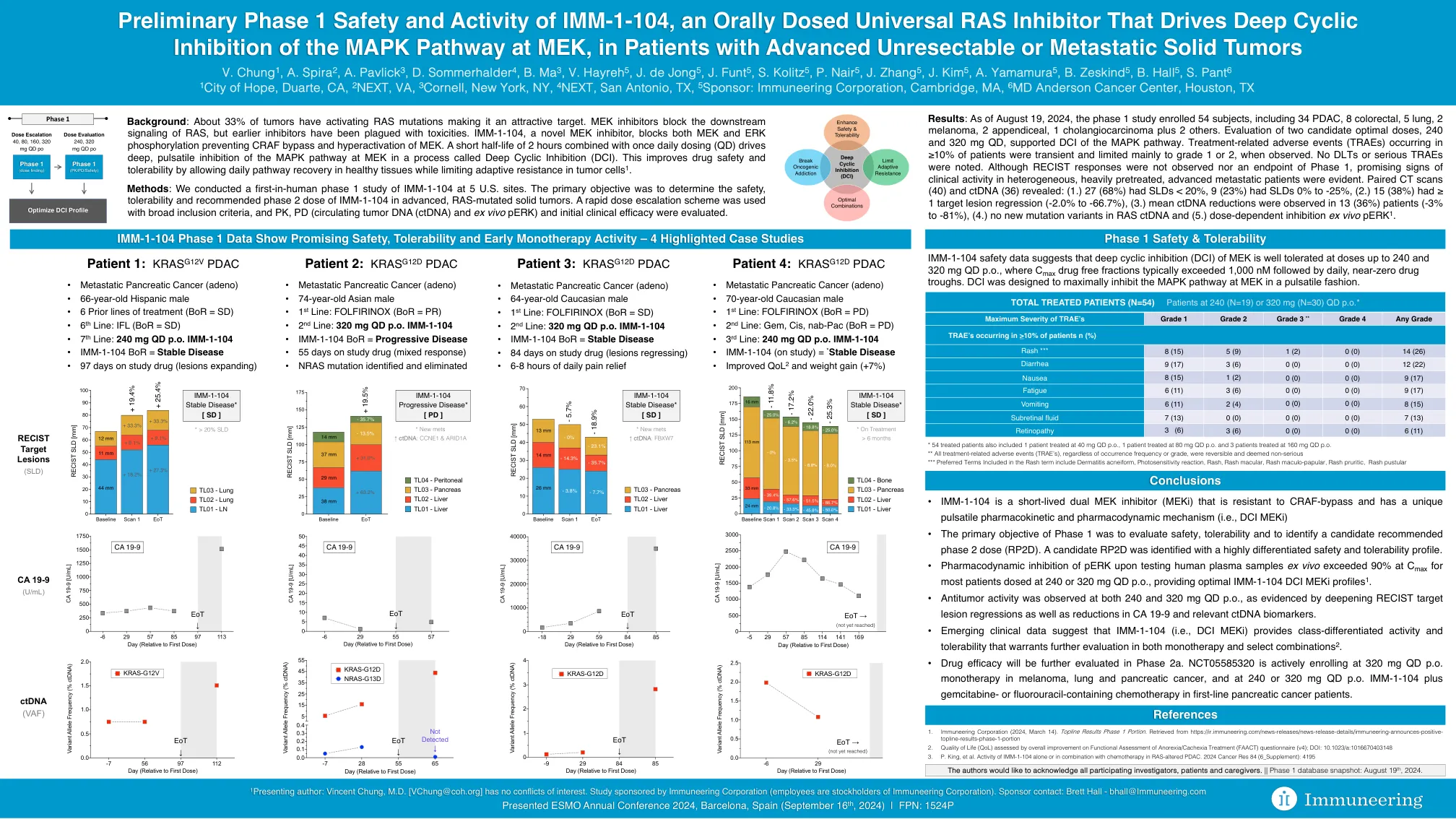
黑色素瘤,2个阑尾,1个胆管癌加2其他。评估两种候选最佳剂量240和320 mg QD,支持MAPK途径的DCI。在≥10%的患者中发生的与治疗相关的不良事件(TRAES)是短暂的,并且在观察到时主要限制为1或2级。没有注意到DLT或严重的Traes。尽管未观察到恢复反应和第1阶段的终点,但在异质,大量预处理的晚期转移性患者中有希望的临床活性迹象。配对的CT扫描(40)和ctDNA(36)显示:(1.)27(68%)的SLD <20%,9(23%)的SLDS为0%至-25%,(2。)15(38%)的目标病变回归(-2.0%至-66.7%),(3。)在13例(36%)患者(-3%至-81%)中观察到平均ctDNA减少。(4。)RAS ctDNA中没有新的突变变体和(5.)剂量依赖性抑制ex Vivo Perk 1。

II型RAF抑制剂ToVorafenib在复发/难治性小儿低级神经胶质瘤中:2期Firefly-1试验
BRAF基因组改变是小儿低级神经胶质瘤(PLGG)中最常见的致癌驱动因素。ARM 1(n = 77)试验研究了口服,选择性,中枢神经系统 - pentrant,II型RAF抑制剂Tovorafenib(420 mg m -2一次,每周420 mg M -2最高600 mg)对BRAF -ARAF -ARETED -ARETED -ARETED -ARTERED -PLACCED的患者的功效。ARM 2(n = 60)是一个延伸队列,它为ARM 1闭合后的RAF改变PLGG患者提供了治疗。基于独立审查,根据神经肿瘤高级神经胶质瘤(RANO-HGG)标准的响应评估,67%的总响应率(ORR)符合ARM 1 1预定的主要端点;响应持续时间(DOR)为16.6个月;响应时间(TTR)的中位时间为3.0个月(次要终点)。通过小儿神经肿瘤学低级神经胶质瘤(RAPNO)标准和安全性评估评估的其他选择的次要终点包括ORR,DOR和TTR(在所有治疗的患者中评估,ARM 2,ARM 2,n = 137)。根据RAPNO标准(包括次要答复)的ORR为51%; DOR中位数为13.8个月; TTR中位数为5.3个月。最常见的治疗不良事件(TRAES)是头发颜色变化(76%),肌酸磷酸激酶(56%)和贫血(49%)。≥3级Traes发生在42%的患者中。九(7%)患者患有TRAES导致停用Tovorafenib。这些数据表明,Tovorafenib可能是对BRAF的,经过重复/难治性的PLGG的有效疗法。临床。GOV注册:NCT04775485。

标题:EZH2抑制剂Tazemetostat Plus Amdizalisib(PI3K抑制剂)的II期研究,患有复发/难治性淋巴瘤的患者
30mg队列的20毫克队列和1分减少了TAZ剂量,来自30mg队列的2分均具有Amdizalisib和Taz剂量降低)。4(36.4%)PTS在30mg队列中经历了TRSAE,而20mg队列的1(10%)PT经历了TRSAE。pts经历了更多的≥3级Traes,trsaes和Teaes,导致剂量

恩科拉非尼联合比尼替尼治疗 BRAFV600 突变转移性非小细胞肺癌患者的 II 期开放标签研究
结果 在数据截止时,98 名患有 BRAF V600E 突变型转移性 NSCLC 的患者(59 名未接受过治疗,39 名曾接受过治疗)接受了 encorafenib 加 binimetinib 治疗。encorafenib 的中位治疗持续时间为 9.2 个月,binimetinib 的中位治疗持续时间为 8.4 个月。未接受过治疗的患者中,按 IRR 计算的 ORR 为 75%(95% CI,62 至 85),曾接受过治疗的患者中为 46%(95% CI,30 至 63);中位 DOR 无法估计(NE;95% CI,23.1 至 NE)和 16.7 个月(95% CI,7.4 至 NE)。24 周后的 DCR 在未接受过治疗的患者中为 64%,在曾接受过治疗的患者中为 41%。初治患者的中位 PFS 为 NE(95% CI,15.7 至 NE),而既往接受过治疗的患者中位 PFS 为 9.3 个月(95% CI,6.2 至 NE)。最常见的治疗相关不良事件 (TRAE) 是恶心 (50%)、腹泻 (43%) 和疲劳 (32%)。TRAE 导致 24 名 (24%) 患者减少剂量,15 名 (15%) 患者永久停用 encorafenib 和 binimetinib。报告了一例 5 级 TRAE 颅内出血。本文中呈现的数据可在 PHAROS 仪表板 ( https://clinical-trials.dimensions.ai/pharos/ ) 上进行交互式可视化。

CX-072(pacmilimab),一种 Probody® PD-L1 抑制剂,...
摘要 背景 Probody ® 治疗药物是抗体前体药物,可在肿瘤微环境中被肿瘤相关蛋白酶激活,从而将活性限制在肿瘤微环境中并最大限度地降低“非肿瘤”毒性。我们报告了 CX-072(pacmilimab)首次人体研究的剂量递增和单药扩增期数据,CX-072 是一种针对程序性死亡配体 1 (PD-L1) 的 Probody 检查点抑制剂。方法 在这项多中心、开放标签研究 (NCT03013491) 的剂量递增阶段,晚期实体瘤成人患者(未使用过程序性死亡-1/PD-L1 或细胞毒性 T 淋巴细胞相关抗原 4 抑制剂)被纳入七个剂量递增队列之一,每 14 天静脉注射一次 pacmilimab。主要终点是安全性和确定最大耐受剂量 (MTD)。在扩展阶段,招募了患有六种预先指定的恶性肿瘤之一(三阴性乳腺癌 [TNBC];肛门鳞状细胞癌 [aSCC];皮肤鳞状细胞癌 [cSCC];未分化多形性肉瘤 [UPS];小肠腺癌 [SBA];和胸腺上皮肿瘤 [TET]);或高肿瘤突变负荷 (hTMB) 肿瘤的患者。主要终点是客观反应(实体肿瘤反应评估标准 v.1.1)。结果剂量高达 30 mg/kg 时未达到 MTD。根据扩展阶段的药代动力学和药效学结果,选择 10 mg/kg 的推荐 2 期剂量 (RP2D)。扩展期招募了 98 名患者:TNBC(n=14)、aSCC(n=14)、cSCC(n=14)、UPS(n=20)、SBA(n=14)、TET(n=8)和 hTMB 肿瘤(n=14)。在 RP2D 接受 pacmilimab 治疗的 114 名患者中,10 名患者(9%)报告了≥3 级治疗相关不良事件 (TRAE),6 名患者(5%)报告了严重 TRAE,2 名患者(2%)因 TRAE 而停止治疗。2 名患者(皮疹、心肌炎)发生了≥3 级免疫相关不良事件。在 22/144 (19%) 名患者中观察到高 PD-L1 表达(即 >50% 肿瘤比例评分)。在患者中观察到了确认的客观反应

e001235.full.pdfCX-072(PACMILIMAB),Probody®PD
抽象的背景探针®治疗药是抗体前药,通过肿瘤相关蛋白酶在肿瘤微环境中激活,从而将活性限制为肿瘤微环境并最小化“非肿瘤”毒性。我们报告了CX-072(PACMilimab)的第一研究剂量 - 定量和单药扩展相位数据,这是针对针对编程死亡配体1(PD-L1)的概率检查点抑制剂。该多中心,开放标签研究(NCT03013491)的剂量升级阶段的方法,具有晚期实体瘤的成年人(天真到程序中降临至dephemed-Death-1/pd-l1或细胞毒性T-淋巴细胞毒性T-淋巴细胞4的抗原4抑制剂)与七剂量施用量施用七剂量的抑制作用。每14天静脉注射一次。主要终点是最大耐受剂量(MTD)的安全性和确定。在扩展阶段,患有六种预先指定恶性肿瘤之一(三阴性乳腺癌[TNBC];肛门鳞状细胞癌[ASCC];皮肤SCC [CSCC];未差异降低的多余型肉瘤[UPS];小肠腺癌[sba]和Thymic thymic [sba];或高肿瘤突变负担(HTMB)肿瘤。主要终点是客观响应(实体瘤的响应评估标准v.1.1)。结果,未达到30 mg/ kg的剂量达到MTD。根据扩展阶段的药代动力学和药效学发现,选择了10 mg/kg的建议2剂量(RP2D)。参加膨胀阶段的九十八名患者:TNBC(n = 14),ASCC(n = 14),CSCC(n = 14),UPS(n = 20),SBA(n = 14),TET(n = 8)和HTMB肿瘤(n = 14)。在RP2D接受PACMILIMAB的114例患者中,有10例患者(9%),6例患者(5%)的严重TRAES(5%)和由于两名患者的TRARES导致治疗停药(2%)(2%),据报道了与治疗相关的不良事件(TRAES)。≥3级与免疫相关的AE发生在两名患者中(皮疹,心肌炎)。高PD-L1表达(即> 50%的肿瘤比例评分)。在患者中观察到确认的客观反应