XiaoMi-AI文件搜索系统
World File Search System突出端

使用 T4 DNA 连接酶 (M0202) 进行连接
3. 对于粘性末端,在 16°C 下孵育过夜或室温下孵育 10 分钟。 4. 对于平末端或单碱基突出端,在 16°C 下孵育过夜或室温下孵育 2 小时(或者,高温孵育)

使用 T4 DNA 连接酶 (M0202) 进行连接
3. 对于粘性末端,在 16°C 下孵育过夜或室温下孵育 10 分钟。 4. 对于平末端或单碱基突出端,在 16°C 下孵育过夜或室温下孵育 2 小时(或者,高温孵育)

蓖麻植物中的 CRISPR/Cas9 介导基因组编辑......
摘要 动机:CRISPR/Cas9 技术已被开发为最有效和最广泛使用的基因组编辑工具,用于修改众多植物的基因组,其中双链 DNA 中的 cas9 切割由单个向导 RNA(sgRNA)中包含的 20 个核苷酸序列驱动。然而,使用 CRISPR/Cas9 同时编辑多个目标仍然是该领域的技术挑战(Ma 等人,2014 年)。方法:在本研究中,使用 Golden Gate Assembly 克隆策略生成多个 CRISPR/cas9 编辑结构以用于蓖麻植物。模块化克隆系统使用 IIS 型酶在其识别位点外切割,从而允许有效组装具有兼容突出端的 DNA 片段,从而同时促进多个序列的正确取向(Engler 等人,2014 年)。我们的主要目标是获得一种遗传构建体,允许在同一个质粒载体中表达两个 sgRNA 和 cas9 核酸酶,以便通过农杆菌感染转化蓖麻。选择了两个针对 FAH12 蓖麻羟化酶的 CRISPR 靶标以避免可能的脱靶。这些靶标包含在 sgRNA 中并克隆到 0 级质粒中,每个质粒两侧都有 BsaI 酶的限制位点。Golden Gate 1 级反应包括几个 BsaI 消化和连接循环,将 U6 启动子与两个 sgRNA 分别组装到 1 级质粒中,两侧都有 BpiI 限制位点。同时,cas9 酶在双强 35S 启动子的控制下克隆,随后是来自 0 级质粒的胭脂碱合酶 (nosT) 终止子,包括这些元素,克隆到另一个 1 级质粒中,两侧也有 BpiI 限制位点。然后,用 BpiI 消化所有 1 级元件(U6-sgRNA1、U6-sgRNA2、2x35S-cas9-nosT)时,会出现兼容的突出端,这些突出端可以以正确的顺序和方向组装成 2 级结构。最终结果是 2 级质粒,其中包括 FAH12 羟化酶的 CRISPR/cas9 多重基因组编辑所需的所有元件。该构建体将转移到农杆菌中,以便以后进行蓖麻胚转化。

癌症中的基因组编辑
基因组编辑是一种利用工程核酸酶在特定基因组位置诱导双链断裂 (DSB) 的方法,以便利用细胞内源性 DNA 修复机制引入基因组修饰 [ 1 , 2 ]。DSB 形成后,细胞将利用两种修复机制中的一种——非同源末端连接 (NHEJ) 和同源性依赖性修复 (HDR),这两种机制均可用于诱导 DNA 变化 [ 3 , 4 ]。在 NHEJ 过程中,细胞将 DNA 的断裂末端重新连接在一起——这个过程很快但往往不准确,修复后的链通常包含小的突变,表现为小的缺失和插入 [ 5 , 6 ]。在基因组编辑中,NHEJ 用于通过功能丧失突变来灭活基因功能。HDR 是一个更复杂的过程,需要供体 DNA 与断裂的两侧都具有同源性。在 HDR 中,细胞处理 DSB 的末端,留下 3′突出端,这些突出端侵入供体 DNA 的同源位点,将其用作 DNA 合成的模板,从而纠正断裂并使其与供体 DNA 相同 [7]。虽然在自然界中,供体 DNA 是姐妹染色单体,但在基因组编辑中,外源 DNA 被引入细胞,作为模板,将所需的变化引入基因组 [8](图 1)。多年来,已有多种类型的工程核酸酶被用于诱导基因组编辑所需的 DSB,包括

利用机器学习预测不同编辑类型和染色质环境下的主要编辑效率
图 1 | 基于序列上下文的 pegRNA 效率表征和预测。(a)使用目标匹配的 pegRNA 文库“Library Diverse”进行筛选的示意图。(b - g)HEK293T 或 K562 中(b,c)插入、(d,e)HEK293T 或 K562 中 1-5bp 替换和(f,g)HEK293T 或 K562 中 1-15 bp 删除的编辑效率。(h,i)在 HEK293T(h)或 K562(i)细胞中安装了 2 个独立 1 bp 替换的双重编辑的编辑效率。预期编辑意味着安装了两个替换,而中间编辑意味着只安装了 2 个替换中的 1 个。距离 0 对应于单个 1 bp 编辑。 (j,k) 在 HEK293T (j) 和 K562 (k) 细胞中,在 GG PAM 序列内进行单 1bp 和双 1bp 替换(有或无编辑)的编辑效率。(d、e、hk) 条形图仅包含具有 7、10 或 15bp RTT 突出端的 pegRNA,以确保不同条件下 RTT 突出端分布相似。(bk) 条形图显示平均值,误差线表示平均值 +/- sem (l、m) PRIDICT2.0 在 Library-Diverse(5 倍交叉验证)上对 (l) HEK293T(n = 22,619)和 (m) K562(n = 22,752)细胞的性能。根据高斯 KDE,颜色渐变从深紫色到黄色表示点密度增加。 (n)PRIDICT2.0 示意图,该模型是基于两个模型的预测平均值的集成模型:(模型 A),

BioXp® Select DNA 克隆试剂盒、Golden Gate 组装体...
图 1. BioXp 上的 Golden Gate 组装。Golden Gate 组装概览。要克隆的插入 DNA 带有侧翼 GG 酶识别位点(BsaI 和/或 BsmBI),可以作为合成基因片段或 PCR 扩增子(1A)和(1B)或预克隆载体格式(1C)获取。用户可以输入任何具有兼容 GG 突出端(以粉色和紫色显示)的所需目标载体(2)。用户在 BioXp 3250 上输入 96 孔板和 GG 克隆条(4)。GG 克隆产品在 BioXp 运行后作为输出交付(5)。

通过 CRISPR 进行高效、安全的基因组编辑...
摘要 CRISPR-Cas12a 系统已被开发用于在真核细胞中实现高度特异性的基因组编辑。鉴于 Cas12a 基因相对较小,该系统被认为最适用于使用 AAV 载体递送的基因治疗。之前,我们报道了富含 U 的 crRNA 能够通过 CRISPR-Cas12a 系统在真核细胞中进行高效的基因组编辑。在本研究中,我们在 crRNA 富含 U 的 3 ′-突出端的核糖 C2 处引入了甲氧基修饰。当与 Cas12a 效应蛋白混合时,核糖基-2 ′-O-甲基化 (2-OM) 富含 U 的 crRNA 能够提高 dsDNA 的消化率。此外,化学修饰的富含 U 的 crRNA 在小鼠受精卵中实现了非常安全且高度特异性的基因组编辑。工程化的 CRISPR-Cas12a 系统有望促进各种动物模型的生成。此外,工程化的 crRNA 也得到了评估,以进一步改进 CRISPR 基因组编辑工具集。

通过单农杆菌系统简化植物基因沉默和基因组编辑物流,同时递送多部分病毒载体
图 1 JoinTRV 的设计,这是一种基于具有兼容来源的微型 T-DNA 载体的 TRV 表达系统。(A) 烟草脆裂病毒 (TRV) 的基因组组织。(B) TRV RNA2 工程用于序列克隆和表达。pLX-TRV2 的克隆盒图与 Bsa I 识别位点和 Bsa I 产生的突出端一起显示(底部)。LacZ 报告基因允许可视化选择重组载体;插入物的植物表达由豌豆早褐病毒 (PEBV) 外壳蛋白 (CP) 启动子驱动。(C) VIGS (pTRV2) 和 VIGE (pRNA2.PEBV) 中描述的 pLX-TRV2 和 TRV RNA2 载体的尺寸比较。(D) JoinTRV 系统图。两个 T-DNA 载体被多路复用到单个农杆菌细胞中,以同时递送 TRV 基因组成分。 pLX-TRV2 是 pLX-B2 衍生物,具有 pBBR1 来源和卡那霉素抗性基因 (npt I),pLX-TRV1 是 pLX-Z4 衍生物,具有 RK2 来源和庆大霉素抗性基因 (aac C1);由于这两个 T-DNA 载体具有兼容的来源和独立的抗生素选择机制,因此可以同时寄宿在同一细菌细胞中
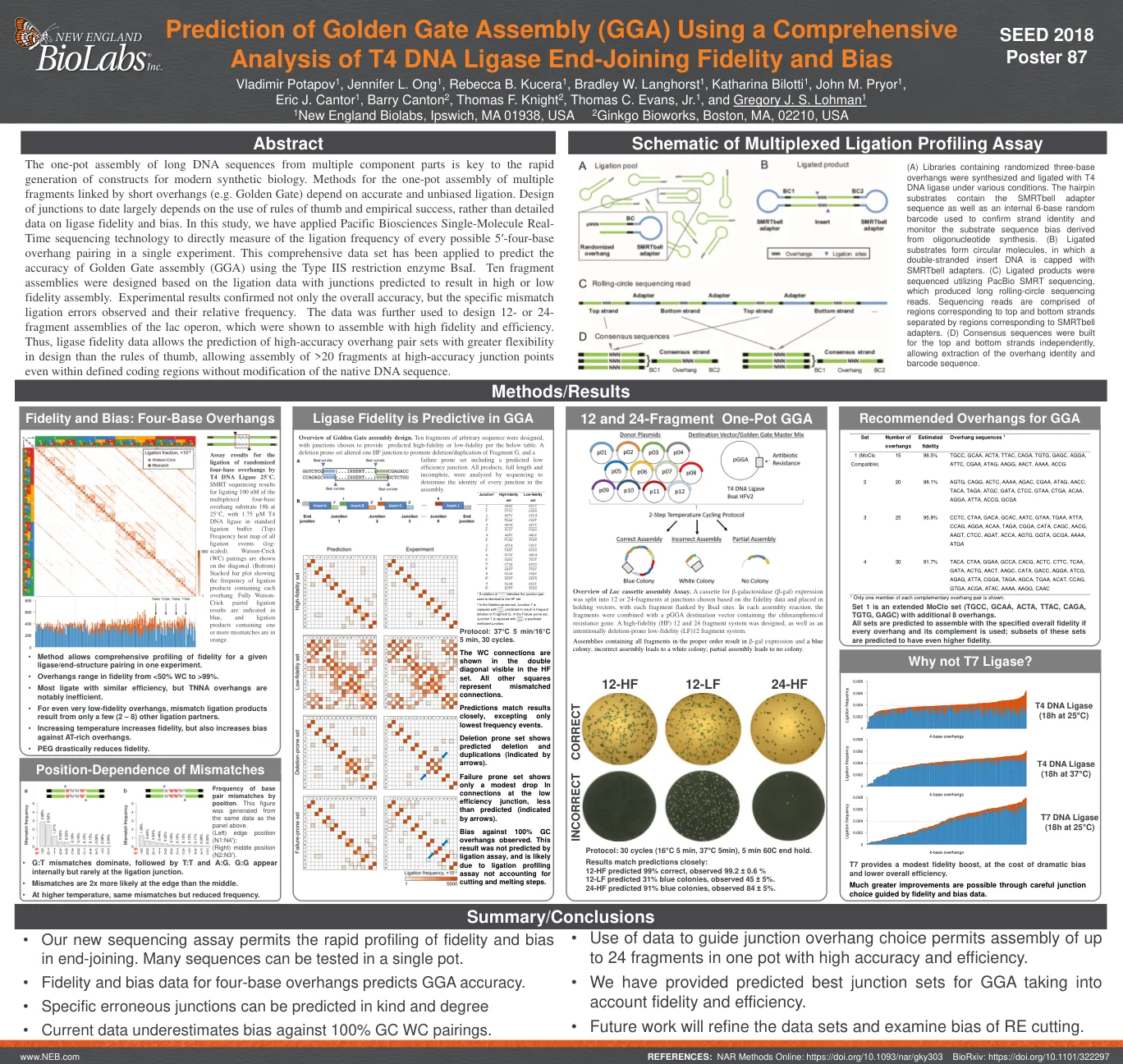
使用综合方法预测金门组装 (GGA)
一锅法组装来自多个组成部分的长 DNA 序列是快速生成现代合成生物学构建体的关键。一锅法组装由短悬垂结构(例如 Golden Gate)连接的多个片段的方法取决于准确和无偏的连接。迄今为止,连接点的设计很大程度上取决于经验法则和经验成功,而不是连接酶保真度和偏向性的详细数据。在本研究中,我们应用 Pacific Biosciences 单分子实时测序技术在一次实验中直接测量每个可能的 5′-四碱基悬垂结构配对的连接频率。该综合数据集已用于预测使用 IIS 型限制性酶 BsaI 的 Golden Gate 组装 (GGA) 的准确性。根据连接数据设计了十个片段组装,其中连接点预测会导致高或低保真度组装。实验结果不仅证实了总体准确性,还证实了观察到的特定错配连接错误及其相对频率。这些数据还用于设计 12 或 24 个片段的乳糖操纵子组装体,结果表明组装体具有高保真度和高效率。因此,连接酶保真度数据可以预测高精度突出端对集,设计灵活性比经验法则更高,即使在定义的编码区域内也可以在高精度连接点组装 20 多个片段,而无需修改天然 DNA 序列。