XiaoMi-AI文件搜索系统
World File Search System统计推断

林昌人 研究兴趣
我的研究领域是信息的数学理论及其应用,特别是研究了通信、统计推断和密码学的数学理论。这些主题有不同的应用方面,并且由于历史原因而具有不同的社区。然而,这些主题具有共同的数学方面。因此,这些主题可以用共同的数学处理方式来处理。我根据共同的数学性质研究了这些主题。具体来说,我主要针对量子系统以及非量子(经典)系统研究这些主题。最近,我用这种方法研究了热力学的基础。最近,我主要在研究以下几点。一是基于群表示理论的量子信息处理的数学处理。群对称性通过消除基依赖性简化了量子系统中的许多问题。事实上,即使给定的信息处理问题由于问题的复杂性而需要进行困难的分析,群对称性也会通过降低复杂性来简化问题。利用群对称性,我们可以构建独立于基的通用协议。由于量子系统的群论方法尚未完成,因此需要进一步发展。第二是信息论保密的数学理论。最近,我为这个主题提出了几种方法,但是它们之间的关系不太清楚,还有一些问题尚未解决。因此,这个主题需要进一步研究。第三是量子理论的基础。虽然以前没有从信息论的角度研究过这个主题,但现在正在从操作的角度用信息论进行研究。我正在研究这个研究方向。

根据机载激光扫描得出的树木直径建模...
过去,在野外准确测量树高比测量树胸高更困难。因此,林业文献中广泛开发了根据直径测量预测树高模型。通过使用机载激光扫描技术(例如 LiDAR),可以准确测量树高和树冠直径等树木变量,这一发展催生了对根据机载激光测量预测直径的模型的需求。尽管已经进行了一些拟合此类模型的工作,但没有一个模型能够结合空间信息来提高预测直径的准确性。使用简单的线性模型,根据激光测得的树高和树冠直径测量结果预测树木直径,我们比较了普通最小二乘法 (OLS)、具有非零相关结构的广义最小二乘法 (GLS)、线性混合效应模型 (LME) 和地理加权回归 (GWR) 的性能。我们的数据来自挪威建立的 36 个样地。这是第一项研究树木级 LiDAR 数据的空间统计模型使用情况的研究。使用 LME 预测树木直径的误差为 3.5%,使用 GWR 预测误差为 10%,使用 OLS 预测误差为 17%。LME 在所有验证类中的预测性能也表现出较低的变异性。考虑到使用参数统计推断(例如基于最大似然的指数)对 GWR 的困难,我们使用置换检验和引导法作为检测统计差异的方法。LME 明显优于其他模型,GWR 优于 OLS 和 GLS。我们的结果表明,LME 模型根据基于 LiDAR 的变量对树木直径的预测效果最佳,达到以前无法达到的程度。
根据机载激光扫描得出的树木直径建模...
过去,在野外准确测量树高比测量树胸高更困难。因此,林业文献中广泛开发了根据直径测量预测树高模型。通过使用机载激光扫描技术(例如 LiDAR),可以准确测量树高和树冠直径等树木变量,这一发展催生了对根据机载激光测量预测直径的模型的需求。尽管已经进行了一些拟合此类模型的工作,但没有一个模型能够结合空间信息来提高预测直径的准确性。使用简单的线性模型,根据激光测得的树高和树冠直径测量结果预测树木直径,我们比较了普通最小二乘法 (OLS)、具有非零相关结构的广义最小二乘法 (GLS)、线性混合效应模型 (LME) 和地理加权回归 (GWR) 的性能。我们的数据来自挪威建立的 36 个样地。这是第一项研究树木级 LiDAR 数据的空间统计模型使用情况的研究。使用 LME 预测树木直径的误差为 3.5%,使用 GWR 预测误差为 10%,使用 OLS 预测误差为 17%。LME 在所有验证类中的预测性能也表现出较低的变异性。考虑到使用参数统计推断(例如基于最大似然的指数)对 GWR 的困难,我们使用置换检验和引导法作为检测统计差异的方法。LME 明显优于其他模型,GWR 优于 OLS 和 GLS。我们的结果表明,LME 模型根据基于 LiDAR 的变量对树木直径的预测效果最佳,达到以前无法达到的程度。

使用唯一分子标识符从下一代测序中改进高分辨率拷贝数变化分析
摘要背景:最近,涉及致癌途径涉及的基因的拷贝数变化(CNV)引起了人们对管理疾病可疑性的越来越多的关注。CNV是肿瘤细胞基因组中最重要的体细胞像差之一。癌基因激活和肿瘤抑制基因失活通常归因于许多癌症类型和阶段的拷贝数增益/扩增或缺失。下一代测序方案的最新进展允许将唯一分子标识符(UMI)添加到每个读取中。每个靶向的DNA片段都用添加到测序引物中的独特随机核苷酸序列标记。umi通过使每个DNA分子在不同的读取群中使每个DNA分子与CNV检测特别有用。结果:在这里,我们提出了分子拷贝数改变(MCNA),这是一种新的甲基动态,允许使用UMI检测拷贝数变化。该算法由四个主要步骤组成:UMI计数矩阵的构建,使用控制样品构建伪参考,log-Ratios的计算,分割以及最后的统计推断异常分段断裂。我们证明了MCNA在患有弥漫性大B细胞淋巴瘤患者的数据集上取得了成功,我们强调MCNA结果与比较基因组杂交具有很强的相关性。结论:我们提供了MCNA,这是一种新的CNV检测方法,可在https:// gitla b.com/pierr ejuli en.viail ly/mcNA/MCNA/MCNA/MCNA/MCNA/MCNA/MCNA/MCNA/MCNA许可下免费获得。MCNA可以通过使用UMI显着提高CNV变化的检测准确性。

神经影像学
典型相关分析 (CCA) 及其正则化版本已在神经影像学界广泛用于揭示两种数据模式(例如,脑成像和行为)之间的多变量关联。然而,这些方法具有固有的局限性:(1)关于关联的统计推断通常不够稳健;(2)未对每种数据模式内的关联进行建模;(3)需要估算或删除缺失值。组因子分析 (GFA) 是一种分层模型,通过提供贝叶斯推断和建模特定于模式的关联来解决前两个限制。在这里,我们提出了一种处理缺失数据的 GFA 扩展,并强调 GFA 可用作预测模型。我们将 GFA 应用于由人类连接组计划 (HCP) 的大脑连接和非成像测量组成的合成和真实数据。在合成数据中,GFA 揭示了潜在的共享和特定因素,并正确预测了完整和不完整数据集中未观察到的数据模式。在 HCP 数据中,我们确定了四个相关的共同因素,捕捉情绪、酒精和药物使用、认知、人口统计和精神病理学测量与默认模式、额顶叶控制、背部和腹侧网络和岛叶之间的关联,以及两个描述大脑连接内关联的因素。此外,GFA 预测了一组来自大脑连接的非成像测量。这些发现在完整和不完整的数据集中都是一致的,并且复制了文献中以前的发现。GFA 是一种很有前途的工具,可用于揭示基准数据集(例如 HCP)中多种数据模式之间的关联,并且可以轻松扩展到更复杂的模型以解决更具挑战性的任务。

用于 NMR 模型推断的量子近似贝叶斯计算
过去几年中,量子技术面临的核心挑战之一是寻找近期量子机器的有用应用 1 。尽管在增加量子比特数量和提高其质量 2、3 方面已经取得了长足的进步,但在不久的将来,我们预计可靠门的数量将受到噪声和退相干的限制——即所谓的嘈杂中尺度量子时代。因此,提出了混合量子-经典方法,以充分利用现有的量子硬件并用经典计算对其进行补充。最值得注意的是,量子近似优化算法(QAOA) 4 和变分量子特征求解器(VQE) 5 的发展。这两种算法都使用量子计算机来准备变分状态,其中一些变分状态可能无法通过经典计算获得,但使用经典计算机来更新变分参数。已经进行了大量实验,证明了这些算法的可行性 6 – 8 ,但它们对现实问题的影响仍不清楚。在基于模型的统计推断中,人们经常面临类似的问题。对于简单模型,可以找到似然值并使其最大化,但对于复杂模型,似然值通常是难以处理的 9,10。NMR 波谱就是一个很好的例子。对于应该使用的模型类型有很好的理解(公式 (1)),人们只需要确定适当的参数。然而,计算特定模型的 NMR 波谱需要在指数级大的希尔伯特空间中执行计算,这对经典计算机来说极具挑战性。这一特性是提出将 NMR 作为量子计算平台的最初动机之一。尽管已经证明 NMR 实验中不存在纠缠 12,13,但强相关性使其在经典上难以处理;也就是说,算子 Schmidt 秩呈指数增长,例如,这禁止有效的表示
WP24 B.Tech。机械工程。 Jntu Hyderabad Page 1 ...
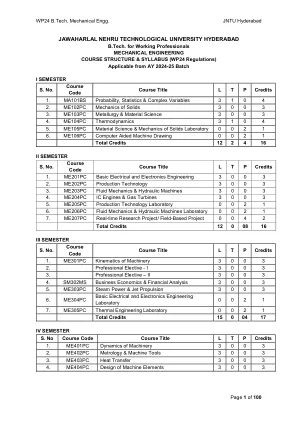
制定和解决涉及随机变量的问题,并应用统计方法来分析实验数据。将假设的估计和检验概念应用于案例研究。参考其分析性,使用Cauchy的积分和残基定理分析复杂函数。Taylor's和Laurent的复杂功能系列扩展。单元I:基本概率8 L概率空间,条件概率,独立事件和Baye定理。Random variables: Discrete and continuous random variables, Expectation of Random Variables, Variance of random variables UNIT-II: Probability distributions 10 L Binomial, Poisson, evaluation of statistical parameters for these distributions, Poisson approximation to the binomial distribution, Continuous random variables and their properties, distribution functions and density functions, Normal and exponential, evaluation of statistical parameters for these distributions单位III:假设的估计和测试10 l引入,统计推断,经典估计方法。:估计点估计值的平均值,标准误差,预测间隔,估计单个样本的比例,两个均值之间的差,两个样本的两个比例之间的差异。统计假设:一般概念,检验统计假设,有关单个均值的测试,对两种均值进行测试,单个比例的测试,两个样本:两倍的测试。教科书:单元-IV:复杂的分化10升限制,复杂函数,分析性,Cauchy-Riemann方程(无证据),找到谐波共轭,基本分析函数(指数,三角学,对数)及其性质及其性质,共形映射,mobius变换。单元V:复杂的集成10 L线积分,库奇定理,库奇的积分公式,分析函数的零,奇异性,泰勒的系列,劳伦特的系列,残基,库奇残基定理(所有定理都没有证明)。

单级篮子试验中的回应率估计
摘要。肿瘤学的治疗进步已基于特定的基因组畸变过渡到靶向治疗。这种转变需要在临床试验中进行创新的统计方法,尤其是在总体协议研究的新兴范式中。篮子试验是一种总体方案,评估了共享共同基因组畸变但在肿瘤组织学上不同的同类群体中的单一治疗方法。在具有运营优势的同时,对篮子试验的分析引入了有关统计推断的挑战。篮子试验可用于确定目标治疗的肿瘤组织学有望足以搬迁以确定临床评估,并可以采用贝叶斯设计来支持这一决策。除了决策之外,对队列特异性响应率的估计对于为后续试验的设计提供了高度相关。这项研究通过仿真研究评估了具有二元结果的七种贝叶斯估计方法,与(频繁的)样本比例估计值形成鲜明对比。目的是提出特定于响应率,重点是平均偏差,平均平方误差和信息借贷程度。探索了各种场景,涵盖了整个队列中的均匀,异类和聚类的响应率。评估方法的性能显示出偏见和精确度的相当大的交易,强调了基于试验特征的方法选择的重要性。Berry的方法在异质性有限的情况下表现出色。在更一般的情况下没有明确的获胜者出现,方法性能受到了对整体平均值,偏见以及先验和调整参数的选择的收缩量所影响。挑战包括方法的计算复杂性,需要仔细调整参数和先前的分布规范以及对其选择的明确指导。研究人员应在设计和分析篮子试验时考虑这些因素。

定量组测试问题的非自适应算法
考虑具有k非零条目的n维二进制特征向量。可以将矢量作为与n个项目有缺陷的n个项目相对应的入射向量。定量组测试(QGT)问题旨在通过查询返回有缺陷项目总数的项目的子集来学习此二进制特征向量。我们在非自适应方案下考虑了这个问题,在非自适应方案中,子集的查询是集体设计的,并且可以并行执行。大多数现有的有效的非自适应算法用于sublerearymemime,其中k“nα具有0×αα1的nα均未与信息理论下限,logk。最近,Hahn-Klimroth和Müller(2022)通过提供了一种非自适应算法,具有O P N 3 Q的解码复杂性,缩小了这一差距。在这项工作中,我们提出了一种串联的施工方法,该方法产生了一种非自适应算法,其解码复杂性的解码复杂性是O p n2α`n log 2 n q。通过建立QGT问题与所谓的球与垃圾箱问题之间的联系来分析解码失败的概率。我们的算法减少了信息理论和计算界的差距,以从日志k到log log k的所需查询/测试数量。这缩小了具有O P N 2 Q解码复杂性的算法类别中非自适应算法的测试数量的差异。关键字:统计推断,定量组测试,urn模型,压缩传感此外,尽管我们的算法在测试数量方面表现出log k差距,但仅在k异常大的α值中,ką1027对于α“ 0.7),仅在k异常大的情况下,它超过了现有的渐近最佳构造,从而突出了我们提议的construction。

教学大纲第三年艾滋病 V 和 VI 学期.pdf
课程目标: 1. 提供有关数据处理的必要知识,并使用统计和机器学习方法对实际问题进行分析 2. 使用编程工具生成报告并以图形形式可视化结果 预期课程成果: 1. 能够获得数据科学的基本知识 2. 将实时数据转换为适合分析的形式 3. 通过统计推断从数据中获取见解 4. 使用机器学习技术开发合适的模型并分析其性能 5. 确定需求并可视化结果 6. 分析模型的性能和结果质量 单元:1 简介 4 小时 数据科学: 数据科学简介 – 数字宇宙 – 数据来源 – 信息共享 – 数据科学项目生命周期: OSEMN 框架 单元:2 数据预处理和概念学习 6 小时 数据预处理简介 – 读取、选择、过滤数据 – 过滤缺失值 – 操作、排序、分组、重新排列、排名数据假设的制定 –概率近似正确学习 - VC 维度 - 假设消除 - 候选消除算法 单元:3 R 基础知识 8 小时 R 基础知识 - 数据类型和对象 - 控制结构 - 数据框 - 特征工程 - 缩放、标签编码和独热编码、缩减 单元:4 使用 R 进行模型拟合 8 小时 回归模型 - 线性和逻辑模型,分类模型 - 决策树、朴素贝叶斯、SVM 和随机森林,聚类模型 - K 均值和层次聚类 单元:5 可视化 6 小时 数据可视化:箱线图、直方图、散点图、热图 - 使用 Tableau - 异常值检测 - 数据平衡 单元:6 R 中的性能评估 4 小时 损失函数和误差:均方误差、均方根误差 - 模型选择和评估标准:准确度、精确度、F1 分数、召回率 - 二元预测分类 - 灵敏度 - 特异性。