XiaoMi-AI文件搜索系统
World File Search System萎缩

对因 TUBB4A 突变导致基底神经节和小脑萎缩、髓鞘形成不足的青少年进行深部脑刺激:示例 c
在出生后的第一年,他的运动和发育里程碑正常,但随后在 18 个月大时出现肌张力低下和平衡问题。他在 5 岁时出现右下肢肌张力障碍,并开始逐渐丧失粗大和精细运动功能,包括独立行走的能力。他在 7 岁时发展为全身性肌张力障碍。考虑手术时的表型表现主要是肌张力过高,四肢活动范围有限,无法负重,脊柱侧弯严重。他的肌张力障碍对肠内苯海索、巴氯芬、卡比多巴-左旋多巴、亚叶酸钙和丁苯那嗪也有抵抗力。根据他父母的报告,在植入一个

等离子体-Secretase1浓度与具有AD的风险的认知健康个体中的基础前脑萎缩和神经变性相关
Andrea Vergallo,Pablo Lemercier,Enrica Cavedo,Simone Lista,Eugeen Vanmechelen等。等离子体ββ-SECRET1 1。 。 。 。 。 。10.1002/alz。
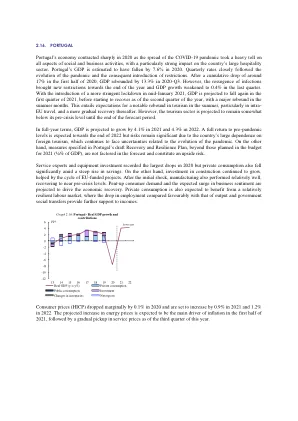
2020 年,由于新冠疫情的蔓延给社会和商业活动的各个方面造成了严重影响,葡萄牙经济急剧萎缩
2020 年,由于新冠疫情的蔓延严重影响了社会和商业活动的方方面面,葡萄牙经济急剧萎缩,对该国庞大的酒店业影响尤为严重。据估计,2020 年葡萄牙的 GDP 下降了 7.6%。季度增长率密切关注疫情的发展和随之而来的限制措施。在 2020 年上半年累计下降约 17% 之后,2020 年第三季度 GDP 反弹 13.3%。然而,感染病例的复发在年底带来了新的限制措施,最后一个季度 GDP 增长放缓至 0.4%。随着 2021 年 1 月中旬实施更严格的封锁措施,预计 GDP 将在 2021 年第一季度再次下降,然后从第二季度开始复苏,夏季几个月出现大幅反弹。这意味着预计夏季旅游业将出现明显反弹,尤其是欧盟内部旅游,此后将逐步复苏。然而,预计旅游业在预测期结束前仍将略低于危机前的水平。

在青少年酒精暴露期间对P75NTR的调节可防止胆碱能神经萎缩和相关的乙酰胆碱活性和行为功能障碍
概念化,BTK和LMS;方法论,BTK和LMS;验证,LMS;正式分析BTK和LMS;调查,BTK和LMS;资源,LMS;数据策划,BTK;撰写第1稿,BTK,写作 - 评论和编辑,LMS;可视化BTK和LMS;监督,LMS; LMS项目管理;资金获取,LMS,BTK

建模大脑萎缩动力学增强了预测阿尔茨海默氏病连续体认知能力下降
阿尔茨海默氏病(AD)被认为是认知能力下降的连续体,而潜在的生物学变化,预测疾病进展至关重要。脑萎缩状态对于评估疾病的严重程度和预后是关键的,但是对其纵向变化及其与进展的统治建模尚未得到充分兴奋。本研究提出了一种基于深度学习的新型方法,可以精确地模拟从ADNI数据库中收集的轻度认知障碍(MCI)受试者的62个皮质和皮质下区域的萎缩动力学,随后是独特的培训方案,以添加β-淀粉样蛋白蛋白质对促性蛋白质的影响以及对模型型的特征的影响。此外,我们将动力学直接实施到MCI中,以将动态实施到示例转换预测任务。我们的发现证明了使用深度学习对萎缩动力学进行建模的可行性,并建议利用动力学代表(DR)增强了转换预测。

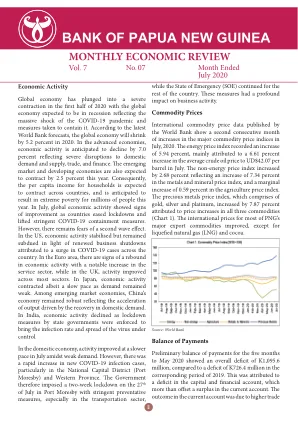
经济活动 全球经济陷入...
2020 年上半年,全球经济陷入严重收缩,预计全球经济将陷入衰退,这反映了新冠疫情大流行的巨大冲击以及为遏制疫情而采取的措施。根据世界银行最新预测,2020 年全球经济将萎缩 5.2%。在发达经济体,预计经济活动将下降 7.0%,反映出国内供需、贸易和金融受到严重破坏。预计新兴市场和发展中经济体今年也将萎缩 2.5%。因此,预计各国家庭人均收入都将萎缩,预计今年将导致数百万人陷入极端贫困。7 月份,随着各国放松封锁并取消严格的新冠疫情遏制措施,全球经济活动显示出改善的迹象。然而,人们仍然担心第二波疫情的爆发。美国经济活动趋于稳定,但因全国新冠病例激增导致企业再次停工,经济活动仍保持低迷。欧元区经济活动出现反弹迹象,服务业显著增长,而英国大部分行业的经济活动均有所改善。日本经济活动萎缩,但速度缓慢,因为需求仍然疲软。在新兴市场经济体中,中国经济保持强劲,反映出国内需求复苏推动产出加速。在印度,由于各邦政府实施封锁措施以控制病毒感染率和传播,经济活动出现下滑。

黎巴嫩经济危机:现状与未来方向
-遗憾的是,当整个世界,特别是我们整个中东和北非地区正在从大流行的萎缩中复苏,并在 2021 年实现健康的正增长率时,黎巴嫩去年却经历了又一年的萎缩,原因是投资几乎缺失(投资总额处于内战以来的最高水平——没有人投资)和实际消费总量疲软(在家庭实际收入急剧下降的情况下),而由于强烈的财政整顿要求和随之而来的紧缩需求,政府支出无法弥补这一损失。

冠状病毒及其对英国经济产出的影响
最近的分析解释了我们分析 GDP 的最新立场,包括我们将如何继续承认“技术性”衰退,即至少连续两个日历季度 GDP 萎缩。虽然这些早期估计仍有修改的可能,但我们更愿意关注因冠状病毒大流行而发生的收缩幅度。很明显,英国正处于有记录以来最大的衰退中。我们最新的估计显示,英国经济现在比 2020 年 2 月萎缩了 17.2%,其影响在那些受公共卫生限制和社交距离影响最大的行业中最为明显。
![神经胶质细胞成像区分多系统萎缩和帕金森病:利用 [ 11 C ] PBR28 和机器学习分析进行正电子发射断层扫描研究](/simg/d\dbe141be97bad77b0a6908bb6f02f4c86eeef9b4.png)
神经胶质细胞成像区分多系统萎缩和帕金森病:利用 [ 11 C ] PBR28 和机器学习分析进行正电子发射断层扫描研究
1 PET 科学中心、个性化医疗和生物样本研发部、阿斯利康公司,瑞典斯德哥尔摩 2 临床神经科学系、精神病学研究中心、卡罗琳斯卡医学院,瑞典斯德哥尔摩 3 陶布研究所、神经病学系、哥伦比亚大学欧文医学中心,美国纽约 4 Invicro,英国伦敦 5 神经影像科学中心、精神病学、心理学和神经科学研究所、伦敦国王学院,英国伦敦 6 耶鲁大学 PET 中心,美国康涅狄格州纽黑文 7 图尔库 PET 中心、图尔库大学和图尔库大学医院,芬兰图尔库 8 研发部、阿斯利康公司,美国马萨诸塞州沃尔瑟姆 9 临床神经科学系、卡罗琳斯卡医学院,瑞典斯德哥尔摩 10 法国 MSA 参考中心、临床研究中心 CIC1436、神经科学和临床药理学系、NeuroToul COEN 中心,UMR 1 214-ToNIC 和图卢兹大学医院、INSERM 和图卢兹 3 大学,法国图卢兹 11 CRMR AMS,神经病学-神经变性疾病服务中心,CHU Bordeaux,法国波尔多 12 波尔多大学,CNRS,IMN,UMR 5293,法国波尔多 13 奥塔哥大学医学系,新西兰基督城脑研究所,新西兰基督城 14 萨勒诺大学神经退行性疾病中心,意大利萨勒诺 15 因斯布鲁克医科大学神经病学系,奥地利因斯布鲁克 16 纽约大学格罗斯曼医学院医学系,美国纽约 17 因斯布鲁克医科大学神经病学系临床神经生物学分部,奥地利因斯布鲁克