XiaoMi-AI文件搜索系统
World File Search System假基因

假基因使基于 Cas9 的基因编辑变得复杂
CRISPR-Cas9 介导的诱导多能干细胞基因编辑成为一种有效的工具,可用于研究遗传驱动疾病的生物学机制,同时考虑各自的遗传背景。该技术依赖于针对目标基因中存在的特定核苷酸序列。因此,某些基因的基因编辑可能因非编码假基因而变得复杂,这些假基因与其各自的基因具有高度的序列同源性。其中,GBA 引起了特别的关注,因为它是帕金森病最常见的遗传风险因素。在本研究中,我们提出了一种易于使用的 CRISPR-Cas9 基因编辑策略,允许对基因中的点突变进行特定编辑,而无需对其假基因进行遗传改变,例如在 GBA 中纠正或插入常见的 N370S 突变。通过结合荧光和 PCR 筛选的质量控制策略,可以早期识别正确编辑的克隆,并明确识别其假基因 GBAP1 的状态。功能验证证实基因编辑成功。我们的工作首次基于 CRISPR-Cas9 对 GBA 中的点突变进行编辑,并为由于存在假基因而技术要求高的基因工程铺平了道路。

BRCA1 假基因通过抑制先天免疫防御机制负面调节抗肿瘤反应
摘要 ◥ 先天免疫防御机制在抗肿瘤反应中起着关键作用。最近的证据表明,抗病毒先天免疫不仅受外源性非自身 RNA 调节,还受宿主衍生的假基因 RNA 调节。越来越多的证据还表明假基因作为基因表达调节剂或免疫调节剂的生物学作用。在这里,我们报告了 BRCA1 肿瘤抑制基因的假基因 BRCA1P1 在调节乳腺癌细胞先天免疫防御机制中的重要作用。BRCA1P1 通过发散转录在乳腺癌细胞中表达长链非编码 RNA (lncRNA)。与正常乳腺组织相比,乳腺肿瘤中 lncRNA-BRCA1P1 的表达增加。BRCA1P1 的消耗会诱导抗病毒防御样程序,包括乳腺癌细胞中抗病毒基因的表达。此外,缺乏 BRCA1P1 的癌细胞通过刺激细胞因子和诱导细胞凋亡来模拟病毒感染的细胞。因此,BRCA1P1 的消耗会增加宿主的先天免疫反应

健康和疾病中的假基因介导的 ceRNA 网络:多维调控视角
缩写:Ψ,假基因;ceRNA,竞争内源性RNA;MRE,微小RNA反应元件;miRNA,微小RNA;TSG,肿瘤抑制基因;mRNA,信使RNA;PP,加工假基因;UP,未加工假基因;UPG,单一假基因,RT,逆转录转座;LINE,长散在核元件;siRNA,短干扰RNA;circRNA,环状RNA;AD,阿尔茨海默病;FTH1,铁蛋白重链;;PTENP1,PTENP1假基因;HMGEC,人乳腺上皮细胞;CRDP,环状RNA衍生的假基因;;HMGA1P,高迁移率族AT-Hook 1假基因;RBP,RNA结合蛋白;;lncRNA,长非编码RNA;CRC,染色质重塑复合物;ERK,细胞外信号调节激酶; BRAF,B-Raf原癌基因;PI3K,磷酸肌醇3-激酶;AKT,丝氨酸/苏氨酸激酶;MAPK,丝裂原活化蛋白激酶;qRT-PCR,定量逆转录聚合酶链反应;FISH,荧光原位杂交;ceRNA假说,竞争性内源性RNA假说;PTPN11,蛋白酪氨酸磷酸酶,非受体型11;NDs,神经退行性疾病;EGFR,上皮生长因子受体;TNF,肿瘤坏死因子;早期生长反应蛋白1(EGR1),HMGA,高迁移率族at-hook 1基因;PMOM,精准医疗肿瘤学市场;scRNA-seq,单细胞RNA测序;ISH,原位杂交;RNAi,RNA干扰;LNP,脂质纳米颗粒; BCL,B 细胞淋巴瘤;AI,人工智能;IP,免疫沉淀;RIP,RNA 免疫沉淀;HRISH,高分辨率原位杂交
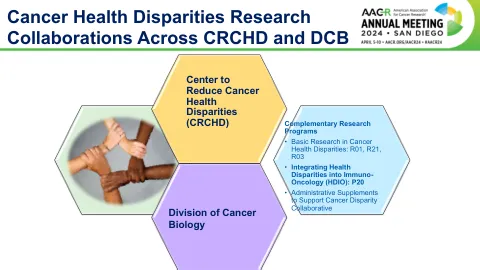
癌症健康差异(CHD)研究
o表征非裔美国人和高加索患者乳腺癌亚型中独特的假基因表达和免疫浸润。免疫相关事件o开发动物模型,这些模型概括了跨不同人群的免疫反应的广度o表征非裔美国人和高加索患者乳腺癌亚型中独特的假基因表达和免疫浸润。免疫相关事件o开发动物模型,这些模型概括了跨不同人群的免疫反应的广度

我的风险+遗传+癌症+技术+规格.pdf
本文提到的所有测试均遵循相同的实验室流程。样本通过基于杂交捕获的靶标富集策略进行制备,以便进行后续的下一代测序。患者基因组 DNA 的等分试样被打碎。通过连接含有独特患者索引的测序接头,将碎裂的 DNA 构建成一个文库。该文库经过纯化,然后通过与一组生物素化探针杂交来富集感兴趣的靶标,然后将其捕获在链霉亲和素包被的珠子上。然后将索引样本汇集并加载到大规模并行的下一代测序仪上进行双端测序。探针设计和 NGS 数据分析针对具有已知假基因区域的基因进行了优化,并根据需要进行了额外的确认测试。

宇宙是不对称的,小鼠大脑也是
图2 - 许多同源基因在蝎子和蜘蛛基因组中保留。a)家族对同源物基因的热图。重复的关系(物种特定于最近的串联重复或家庭/物种特定的损失除外)。o:保留了ohnologue; SOL:Spider Ohnologue失去了; EOL:Entelegyne Ohnologue失去了; AT:古代串联复制; RT:最近的串联重复; SC:保留单副本。*D. plantarius和P. tepidariorum中的第二个FTZ OHNOLOGUS可能是假基因,因为同源域中有停止密码子。b)所包含的节肢动物谱系中古代和最新串联重复的推断时间。基因家族在分支上面列出(以及graminicola H. graminicola)。物种缩写如表2所示。

用BATH对蛋白质编码DNA的敏感且耐误差的注释
我们提出了Bath,这是一种基于该DNA与蛋白质序列数据库的直接比对或对蛋白质序列的数据库的直接比对或蛋白质序列或profe file file file隐藏的马尔可夫模型(PHMMS)的高度敏感注释的工具。BATH建立在HMMER3代码库的顶部,并通过提供直接的输入接口和易于解释的输出来简化基于PHMM的注释的注释工作。BATH还引入了新型的Frameshift感知算法,以检测诱导核苷酸插入和缺失(Indels)。BATH匹配HM-MER3对于包含误差的序列注释的准确性,并产生与所有经过测试的工具相比,用于含有核苷酸indels的序列的所有测试工具。这些结果表明,当需要高注释灵敏度时,应使用浴缸,尤其是当预期的移码误差被期望中断蛋白质编码区域时,与长期读取的数据和假基因的背景下一样。

真核生物核糖体中药物结合残基的自然变异
标题 真核核糖体中药物结合残基的天然变异 作者 Lewis I. Chan 1,& 、Chinenye L. Ekemezie 1,& 、Karla Helena-Bueno 1 、Charlotte R. Brown 1 、Tom A. Williams 2,* Sergey V. Melnikov 1,* 附属机构 1 纽卡斯尔大学生物科学研究所,英国泰恩河畔纽卡斯尔,NE2 4HH 2 布里斯托尔大学生物科学学院,英国布里斯托尔,BS8 1TQ & 贡献相同 通讯 * 通讯地址:tom.a.williams@bristol.ac.uk 和 sergey.melnikov@newcastle.ac.uk 摘要 针对真核核糖体的药物作为研究工具和针对癌症、真菌和其他致病性的潜在疗法正变得越来越重要真核生物。然而,由于缺乏比较研究,我们目前不知道有多少真核生物拥有与人类相同的核糖体药物结合位点,以及有多少与人类有显著差异。目前,这种知识上的差距因真核生物基因组中存在假基因而加剧,由于我们无法区分真正的突变、假基因和测序伪影,使得这些比较分析具有挑战性。在本研究中,我们通过使用一种利用物种间进化关系的新方法解决了这个问题。使用这种方法,我们确定了 8,563 种代表性真核生物中 58 种核糖体药物结合残基的序列变体,追溯了这些变异的进化历史,从 20 亿年前真核生物的出现到它们随后分化成不同的谱系。出乎意料的是,我们发现酵母和人类(通常用作研究核糖体/药物相互作用的模型真核生物)与大多数其他真核生物不同,因为 rRNA 替换主要发生在动物和真菌中,但在大多数其他真核生物中不存在。此外,我们证明了以前在常见病原体利什曼原虫和疟原虫中发现的结构变异,这些变异被视为少数真核生物物种所特有的,但实际上为大量真核生物所共有。值得注意的是,一些真核生物谱系的核糖体药物结合位点与人类的差异比人类与细菌的差异更大。总体而言,我们的研究提供了真核生物核糖体药物结合位点进化的最完整概述(在单个物种、单个残基和单个药物的水平上),确定了与人类相比具有结构不同的核糖体药物结合位点的真核生物谱系。这些发现为利用核糖体靶向药物作为研究工具和开发针对真核寄生虫的谱系特异性抑制剂开辟了新的途径。

非编码RNA在结直肠癌中的作用
摘要:结直肠癌是全球最常见的癌症死亡原因之一。尽管近年来对结直肠癌发病分子机制的认识不断进步以及更有效的药物治疗的实施,但患者的总体生存率仍然不令人满意。高死亡率主要是因为大约一半的癌症患者发生癌症转移以及癌细胞耐药群的出现。对癌症分子生物学认识的不断提高突出了非编码RNA(ncRNA)在结直肠癌发展和进化中的作用。ncRNA通过多种机制调控基因表达,包括表观遗传修饰和长链非编码RNA(lncRNA)与微小RNA(miRNA)和蛋白质的相互作用,以及通过lncRNA作为miRNA前体或假基因的作用。 LncRNA 也可在血液中检测到,循环 ncRNA 已成为结直肠癌诊断和预后以及预测药物治疗反应的非侵入性癌症生物标志物的新来源。在本综述中,我们重点关注 lncRNA 在结直肠癌发展、进展和化学耐药性中的作用,以及作为可能的治疗靶点。
Jumpcode Genomics海报模板
下一代测序(NGS)基于靶向基因面板,外显子组测序(ES)和基因组测序(GS)现在通常用于询问大量基因以供诊断使用。否则对所选的测定法,具有高序列同源性的基因仍然是短读技术的主要挑战,并且可能导致假阳性和假阴性诊断错误。长阅读测序有可能解决许多基因的问题,但是对同源序列的分析通常需要先进的生物信息学管道,这些管道尚未验证用于临床使用。传统上,实验室使用了靶向的Sanger测序和/或远程PCR技术来解决这些基因。这些方法是基因特异性的,难以设计,并且在临床环境中执行昂贵。具有同源性的目标区域是复杂的,可以通过不同程度的同源性,医学相关性和同源性类型(功能同源性,已知的假基因,部分或基因同源性,未表征的非编码区域)进行划分。为了满足这种未满足的需求,我们描述了使用CRISPRCHEAN®技术的概念证明,该技术利用CRISPR-CAS9系统的特异性来降低了下一代测序库中丰富的,无信息的序列。