XiaoMi-AI文件搜索系统
World File Search System同工酶

etoricoxib -FDA验证门户
药效和药代动力学:eToricoxib是COX-2选择性抑制剂。它有选择地抑制环氧酶酶(COX-2)的同工型2。这减少了蛛网膜酸的前列腺素(PGS)的产生。在PG所发挥的不同功能中,应突出显示其在炎症级联反应中的作用。cox-2选择性抑制剂(又称“ coxib)在1型环氧酶中表现出明显的活性。依他氧化的吸收率是中等的,当给予口服时,最大的等离子药物浓度在大约1小时后发生与范围和范围相似etoricoxib广泛结合,主要与血浆白蛋白结合,并且在人类中的分布明显为120 l。与口服剂量在5至120 mg之间增加的血浆浓度时间曲线(AUC)下的面积相比增加。在健康受试者中,消除大约20小时的半衰期可以使每天一次给药。etorigoxib,这些代谢物在尿液和粪便中排泄,而在尿液中很少消除药物(<1%)。etoricoxib主要由细胞色素P450(CYP)3A4同工酶代谢。

用于药物安全相关分类任务的机器学习模型
摘要 在本综述中,我们概述了过去六年(2015-2021 年)与 ADME(吸收、分布、代谢和排泄)和毒性终点相关的机器学习驱动分类研究领域的最新趋势。该研究仅关注具有大型数据集(即超过一千种化合物)的分类模型。针对九个不同的目标进行了全面的文献检索和荟萃分析:hERG 介导的心脏毒性、血脑屏障穿透、通透性糖蛋白 (P-gp) 底物/抑制剂、细胞色素 P450 酶家族、急性口服毒性、致突变性、致癌性、呼吸毒性和刺激/腐蚀。最佳分类模型的比较旨在揭示机器学习算法和建模类型、端点特定性能、数据集大小和不同验证协议之间的差异。根据对数据的评估,我们可以说基于树的算法(仍然)占据主导地位,共识建模在药物安全性预测中正成为一种日益增长的趋势。尽管人们已经可以找到对 hERG 介导的心脏毒性和细胞色素 P450 酶家族同工酶表现出色的分类模型,但这些目标仍然是 ADMET 相关研究工作的核心。

GSTM1*0和CYP1A1*2A(rs4646903)变体的流行率:4p Medicine的预测遗传生物标志物的试验研究
CYP1A1同工酶负责将procarcinogen的生物转化,例如苯并(a)pyrene,纳入反应性化合物。同时,GSTM1通过与谷胱甘肽结合来促进这些代谢产物的排毒。CYP1A1*2A遗传变异的存在加强了这些反应性代谢物的产生,而GSTM1基因的缺失(GSTM1*0)损害了它们的排毒。这种酶促失衡会导致DNA加合物的形成,众所周知,这些加合物会为癌症和其他疾病贡献。鉴于在4P药物框架内研究这些基因的重要性(预测性,预防性,个性化和参与性),这项研究的主要目的是研究秘鲁中部沿海人口中GSTM1*0和CYP1A1*2A的普遍存在。该研究包括秘鲁城镇ICA和利马城镇的131个个人居民。结果显示GSTM1*0的频率为0.47,CYP1A1*2A的等位基因频率为0.68。CYP1A1*2A的基因型频率为6%*1A/*1A,53%*1A/*2A和41%*2a/*2a。值得注意的是,CYP1A1的人口样本不在耐寒的韦恩伯格平衡中(χ2= 5.324)。本研究中报道的GSTM1*0和CYP1A1*2A的频率与先前记录的其他拉丁美洲和三角洲人群的频率不同,可能反映了独特的

肽抑制剂和荧光探针,用于选择性抑制和标记第XIIIA转谷氨酰胺酶
摘要:第XIIIA(FXIIIA)是一种主要治疗兴趣的转谷氨酰胺酶,这是由于其在血液凝结级联反应中的重要作用而发展抗凝剂。虽然已经报道了许多FXIIIA抑制剂,但由于缺乏代谢稳定性和对转谷氨酰胺酶2(TG2)的选择性低,因此未能达到临床评估。此外,用于研究FXIIIA活性和定位的化学工具非常有限。为了消除这些缺点,我们设计,合成和评估了21个新型FXIIIA抑制剂的库。亲电战争头,接头长度和疏水单位在小分子和肽支架上有所不同,以优化同工酶的选择性和效力。然后,将先前报道的FXIIIA抑制剂改编成具有若丹明B部分的探针设计,从而产生创新的KM93作为首次已知的溶液探针,旨在选择性地标记具有较高功能的活性FXIIIA(k Inact / k Inact / k I = 127,300 m-1-1-1-5-5-5)。探针KM93在骨髓巨噬细胞中促进了荧光显微镜研究,在细胞培养中标记具有较高效率和选择性的FXIIIA。这些新型抑制剂和探针的结构 - 活动趋势将有助于对活动,抑制和定位FXIIA的未来研究。

全基因组碱基编辑器筛选鉴定酵母中蛋白质丰度的调节剂
摘要 蛋白质是细胞中的关键分子,其丰度不仅在基因表达水平而且在转录后水平受到广泛调控。在这里,我们描述了一种酵母基因筛选方法,该方法能够系统地表征蛋白质丰度调控在基因组中的编码方式。该筛选方法结合了 CRISPR/Cas9 碱基编辑器来引入点突变,并对内源性蛋白质进行荧光标记以方便流式细胞仪读数。我们首先使用单个 gRNA 以及正向和负向选择筛选对酵母中的碱基编辑器性能进行了基准测试。然后,我们研究了 16,452 种基因扰动对代表各种细胞功能的 11 种蛋白质丰度的影响。我们发现了数百种调控关系,包括 GAPDH 同工酶 Tdh1/2/3 与 Ras/PKA 通路之间的新联系。许多已识别的调节因子特定于这 11 种蛋白质中的一种,但我们还发现了一些基因,这些基因在受到扰动时会影响大多数测试蛋白质的丰度。虽然更具体的调控因子通常作用于转录,但广泛的调控因子往往在蛋白质翻译中发挥作用。总的来说,我们的新筛选方法为蛋白质调控网络的组成部分、规模和连通性提供了前所未有的见解。

PARG 敲除 MDA-MB-231 细胞系
描述 PARG 敲除 MDA-MB-231 细胞系是一种 MDA-MB-231 细胞系,其中人类 PARG(聚 ADP-核糖糖水解酶)长同工酶(PARG111、PARG102 和 PARG99)已使用 CRISPR/Cas9 基因组编辑从基因上去除,慢病毒编码 CRISPR/Cas9 基因和针对人类 PARG 的 sgRNA(单向导 RNA)。该细胞系已通过基因组测序和蛋白质印迹分析验证。背景聚(ADP-核糖)糖水解酶 (PARG) 是一种分解代谢酶,参与 PARylated 链的降解,释放 ADP-核糖和寡(ADP-核糖)链。 PAR(聚 ADP 核糖基化)稳态由 PAR 聚合酶 (PARP) 家族和 PARG 调节,以响应细胞应激条件,例如 DNA 损伤反应 (DDR)。PARG 活性与炎症、缺血、中风和癌症中的细胞反应有关。PARG 在乳腺癌中过度表达,与肿瘤生长和存活有关。PARG 活性降低可以增强当前癌症疗法(例如化疗和放疗)的效果,使 PARG 选择性抑制剂抑制成为癌症和免疫疗法中一种有前途的方法。MDA-MB-231 是一种源自乳腺腺癌的上皮细胞系,具有突变的 p53。它是 ER(雌激素受体)、HER2 和 E-钙粘蛋白阴性,用作晚期乳腺癌的模型。应用

细菌CRISPR/CAS9系统作为所有健康问题的有希望的解决方案,并进步的生物工程
缩写:BP1,肿瘤抑制剂p53结合蛋白1; BRCA,乳腺癌抗原;汽车,嵌合抗原受体; CAS9,CRISPR相关蛋白9;级联,抗病毒防御的CRISPR综合体; CMR,CAS模块坡道(重复相关的神秘蛋白质); CMR III-B,多个亚基III型B CRISPR RNA-CAS蛋白; CPF1,Prevotella和Francisella1的CRISPR; CRISPR,定期间隔间隔室; Crrna,Crispr RNA; CSM III-A,多支亚基III-A CRISPR-CAS蛋白; dcas9/ sgrna-sg I,停用cas9/短指南RNA-Sybrr-green i; DNA-PK,DNA-蛋白K; DNA-PKC,DNA蛋白K催化亚基; DSB,双链断裂; ege,额外的基因元素; GRNA,导向RNA; HDR,同源性维修; IAP,碱性磷酸酶同工酶; MRE 11,减数分裂重组11; NHEJ,非同理结局加入; PAM,原始间隔者相邻基序; PD,程序性细胞死亡; RAD,重组酶A;代表,重复的外部回文; RPA,复制蛋白A; RT,逆转录酶; Sgrna,简短的指南RNA; SSB,单链断裂; tracrrna,反式激活CRISPR RNA; XLF,类似XRCC4的因子; XRCC 4,X射线修复交叉补充蛋白4; Yoyo-1,(恶唑黄色)

α-淀粉酶抑制剂筛选试剂盒(比色)
引入α-淀粉酶(EC 3.2.1.1)是消化酶,催化淀粉中内部α-1,4-糖苷链接的水解至较低的分子量产物,例如葡萄糖,麦芽糖和麦芽糖三糖单位。人α-淀粉酶主要在唾液腺和胰腺中表达。这两个同工酶在其催化结构域中具有高度的原发性氨基酸序列相似性(97%)和92%。此外,两种α-淀粉酶在与多克隆抗体的反应上都是免疫学上相同的,具有相同的作用方式,首选底物,是氯激活的,并且在相似的pH值下达到最大活性。在功能上,当用各种底物测试时,它们具有相似但不相同的切割模式。调节α-淀粉酶活性会影响碳水化合物作为能源的利用。 该酶负责人类复杂碳水化合物的分解。 因此,抑制α-淀粉酶可以被视为治疗与碳水化合物摄取有关的疾病的策略,例如糖尿病,肥胖,牙齿腔和牙周疾病。 α-淀粉酶抑制剂筛选试剂盒(比色)(AB283391)可用于筛选/表征α-淀粉酶的潜在抑制剂。调节α-淀粉酶活性会影响碳水化合物作为能源的利用。该酶负责人类复杂碳水化合物的分解。因此,抑制α-淀粉酶可以被视为治疗与碳水化合物摄取有关的疾病的策略,例如糖尿病,肥胖,牙齿腔和牙周疾病。α-淀粉酶抑制剂筛选试剂盒(比色)(AB283391)可用于筛选/表征α-淀粉酶的潜在抑制剂。
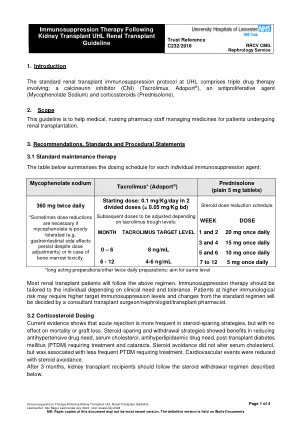
肾移植后UHL肾移植后的免疫抑制疗法
*谨慎 *寻求有关肾脏药房与免疫抑制药物相互作用的建议。西洛里木斯,克莫司和环孢菌素被肠壁和肝脏中的CYP3A4同工酶广泛代谢。CYP3A4的抑制剂(例如酮康唑,伏立康唑,Itraconazole或Clarithromycin)会降低其新陈代谢并增加他克莫司,西洛莫司和ciclosporin水平。 CYP3A4(例如利福平)的诱导剂会增加其新陈代谢并降低他克莫司,西洛里莫斯和ciclosporin水平。 不建议使用与CYP3A4的强抑制剂或CYP3A4诱导剂的强烈抑制剂共同给药。 这不是全面列表,有关互动的更多信息,请联系专业药剂师以寻求建议。CYP3A4的抑制剂(例如酮康唑,伏立康唑,Itraconazole或Clarithromycin)会降低其新陈代谢并增加他克莫司,西洛莫司和ciclosporin水平。CYP3A4(例如利福平)的诱导剂会增加其新陈代谢并降低他克莫司,西洛里莫斯和ciclosporin水平。 不建议使用与CYP3A4的强抑制剂或CYP3A4诱导剂的强烈抑制剂共同给药。 这不是全面列表,有关互动的更多信息,请联系专业药剂师以寻求建议。CYP3A4(例如利福平)的诱导剂会增加其新陈代谢并降低他克莫司,西洛里莫斯和ciclosporin水平。与CYP3A4的强抑制剂或CYP3A4诱导剂的强烈抑制剂共同给药。这不是全面列表,有关互动的更多信息,请联系专业药剂师以寻求建议。

药效团
炎症是影响全球超过 15 亿人的严重公共卫生问题 [1]。其症状包括发热、疼痛、发红、肿胀和功能丧失 [2]。炎症与许多慢性疾病有关,例如糖尿病、癌症、心血管疾病、呼吸系统疾病和自身免疫性疾病 [3-6]。这些使人衰弱的疾病会对患者的生活质量产生重大影响 [7, 8]。抗炎药物的几种作用机制之一是抑制花生四烯酸代谢,该代谢由环氧合酶 (COX) 酶介导,特别是 COX-1 和 COX-2 [9-12]。这两种同工酶的序列几乎相同,唯一的不同之处在于 COX-1 中 523 位的异亮氨酸被 COX-2 中的缬氨酸取代 [13]。异亮氨酸比缬氨酸大,因此可以阻止体积较大的分子(容易与 COX-2 结合)进入 COX-1 的空间位阻侧结合口袋。COX-1 是一种组成酶 [14],对维持组织稳态至关重要,尤其负责产生保护胃内层的天然粘液层 [15, 16]。抑制 COX-1 的药物可能会产生不良反应,例如胃溃疡,这是由于胃中细胞保护性前列腺素的产生减少所致。相反,可诱导的 COX-2 [14] 仅在炎症细胞中表达。因此,那些选择性作用于 COX-2 的药物不会引起与 COX-1 抑制相关的副作用 [17]。传统的 NSAID 是非选择性的;也就是说,它们通过抑制 COX-1 和 COX-2 的活性起作用。较新的 NSAID,特别是所谓的“昔布类”[18-20],对 COX-2 具有显著的选择性。一般来说,市场上现有的 NSAID 具有一系列特定于特定药物的不良副作用 [21, 22]。因此,发现副作用最小或轻微的新型抗炎化合物仍然是一个活跃的研究领域。药物发现中的一种谨慎技术涉及根据已知活性化合物设计或发现新的化学结构。它需要开发作为分子特性函数的生物活性定量模型。