XiaoMi-AI文件搜索系统
World File Search System引物

图S1。 GADD45β和... 的mRNA表达水平 osimertinib诱导的红细胞毛:病例报告 用BNT162B2 ... 对IB3-1细胞的数据S1处理 用软通道微创的激光定位... 表Si。逆转录中使用的底漆序列 - 表Si.定量RT-PCR的序列序列。 自噬/脂肪在响应中的作用... 表Si。研究中使用的siRNA和定量PCR引物序列的列表。 上调和抑制... 的核易位 由于... ,子宫后部区域的脓肿 表Si。逆转录的底漆序列 - 逆转癌症中的DNA高甲基化(综述) 表Si。乳腺癌免疫疗法及其结果的完整临床研究清单。 1个表Si。逆转录中使用的引物 - 3')鼠标-GAPDH f
摘要。本研究报告了osimertinib诱导的转移性肺腺癌Noma患者的红细胞毛的病例。osimertinib是一种通过与细胞内受体酪氨酸激酶位点结合而不可逆地抑制表皮生长因子受体(EGFR)途径的抗肿瘤药物,从而防止EGFR信号转导。伴有肺腺癌复发的77岁女性,并开了次生转移的情况下。患者在两只手的所有手指的远端面积上都表现出痛苦的红斑和温暖,这两只手的远处是热量变质并因冷而缓解。基于临床数据,红细胞毛的肌肌张力。考虑到发病的年龄,排除了原发性红细胞。进一步的研究排除了红细胞毛的其他次要原因,因此怀疑奥西司他尼是原因。尽管尚未报告EGFR抑制剂诱导的红细胞毛的病例,但已记录了EGFR抑制剂引起的皮肤不良事件。目前的情况可能是osimertinib诱导的红血病的第一个证据,可以帮助临床医生正确地支持发展此EGFR抑制剂不良事件的患者。

非瓦里奥拉正oxoxvirus实时PCR引物和探针集-EUA摘要
结果用于鉴定非瓦里奥拉正托病毒DNA。该测定法不会区分该测定法检测到的其他正托病毒,并未检测到Variola病毒。在感染的急性阶段,在人脓疱或囊泡皮疹标本或病毒细胞培养物中,通常可以检测到非瓦里奥拉正oxoxvirus DNA。请参阅CDC算法,急性,广义囊泡或脓疱性皮疹疾病测试方案,并评估患者的天花:急性,全身性囊泡或脓疱性皮疹疾病方案,用于针对患有急性,全身性胸骨或叶面疾病的患者的建议测试和评估算法。该测定的结果是用于鉴定非瓦里奥拉正托病毒DNA。这些结果必须与其他诊断测定法和临床观察结果一起使用,以诊断正托细胞病毒感染。

鼠标引物 - 基因名称
免疫系统是为了抵消不可预测的威胁,但它依靠可预测的活动周期来正常运行。免疫功能中的每日节奏是一个不断扩展的研究领域,许多人源自基于遗传的计时机制,称为昼夜节律。挑战是如何利用这些生物节奏来改善医疗干预措施。在这里,我们回顾了最近的文献,记录了昼夜节律如何组织基本的先天和适应性免疫活动,昼夜节律的免疫学后果和睡眠破坏以及该领域的知识差距。然后,我们考虑将昼夜节律与疫苗接种联系起来的证据,这是免疫功能的重要临床实现。最后,我们讨论了将昼夜节律免疫转换为患者床边的实用步骤。

开发龟环境DNA分析技术 - 通用引物和...
<目的是>〇[实验1]我们将测试不使用玻璃过滤器或碱性的高速离心分离方法,该方法被设计为环境DNA分离方法,并验证其有用性。 〇[实验2]将验证为海龟设计的通用底漆的性能,并将验证其在环境DNA中的使用。 〇[实验3]创建每个鉴定底漆,包括日本乌龟和日本乌龟,并验证其有效性。 〇[实验4]创建软壳龟识别引物并验证其有效性。

通过 DNA 修复调节和 pegRNA 工程改进基于核酸酶的引物编辑 Panagiotis Antoniou* 1、Louis Dacquay* 1,2、Niklas Selfjord 1
通过 DNA 修复调节和 pegRNA 工程改进基于核酸酶的引物编辑 Panagiotis Antoniou* 1 、Louis Dacquay* 1,2 、Niklas Selfjord 1 、Katja Madeyski-Bengtson 3 、Anna-Lena Loyd 3 、Euan Gordon 4 、George Thom 5 、Pei-Pei Hsieh 1 、Sandra Wimberger 1 、Saša Šviković 1 、Mike Firth 6 、Nina Akrap 1 、Marcello Maresca 1# 和 Martin Peterka 1# 1 基因组工程,Discovery Sciences,研发部,阿斯利康,瑞典哥德堡 2 Promega Corporation,美国威斯康星州麦迪逊 3 转化基因组学,Discovery Sciences,研发部,阿斯利康,瑞典哥德堡 4 发现生物学,Discovery Sciences,研发部,阿斯利康,瑞典哥德堡 5 英国剑桥阿斯利康公司发现科学研发部体内表达生物制剂 6 英国剑桥阿斯利康公司发现科学研发部数据科学与定量生物学 * 这些作者的贡献相同 # 通信地址:marcello.maresca@astrazeneca.com martin.peterka@astrazeneca.com
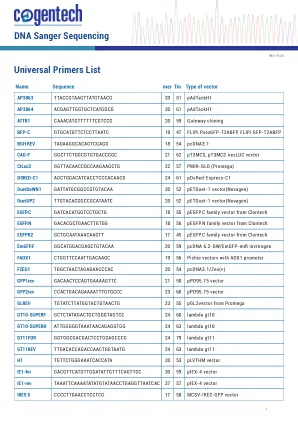
通用引物列表DNA Sanger测序
Cogentech S.R.L.福利公司的成员属于IFOM的管理和协调 - 分子肿瘤学基金会通过Adamello 16,20139年意大利米兰 - 股本€1,100,000 i.v.本地单位:C/O SICILIL S.C.P.A.的科学和技术公园-Z.I。 Palma I Block,V。Lancia,57-95121 Catania P. Iva,C。F。和在米兰公司,Monza,Brianza和Lodi的公司登记册中注册。 04641450962 -R.E.A. MI-1763886 www.cugentech.it |序列service@cogentech.it | ph。 +39 02 57430 3210/3207-Z.I。Palma I Block,V。Lancia,57-95121 Catania P. Iva,C。F。和在米兰公司,Monza,Brianza和Lodi的公司登记册中注册。 04641450962 -R.E.A.MI-1763886 www.cugentech.it |序列service@cogentech.it | ph。+39 02 57430 3210/3207


mRNA的表观遗传调节介导表型... 图S1。 GADD45β和... 的mRNA表达水平 osimertinib诱导的红细胞毛:病例报告 用BNT162B2 ... 对IB3-1细胞的数据S1处理 用软通道微创的激光定位... 表Si。逆转录中使用的底漆序列 - 表Si.定量RT-PCR的序列序列。 自噬/脂肪在响应中的作用... 表Si。研究中使用的siRNA和定量PCR引物序列的列表。 上调和抑制... 的核易位 由于... ,子宫后部区域的脓肿 表Si。逆转录的底漆序列 - 逆转癌症中的DNA高甲基化(综述) 表Si。乳腺癌免疫疗法及其结果的完整临床研究清单。 1个表Si。逆转录中使用的引物 - 3')鼠标-GAPDH f
摘要。可塑性,癌细胞在没有基因组改变的分化状态之间过渡的能力已被认为是肿瘤内异质性的主要来源。它在癌症转移和耐药性中具有至关重要的作用。因此,靶向可塑性具有巨大的希望。然而,癌细胞中可塑性的分子机制仍然鲜为人知。几项研究发现,mRNA充当连接DNA和蛋白质遗传信息的桥梁,在将基因型转化为表型中具有重要作用。本综述概述了通过变化和编辑mRNA进行的调节癌细胞可塑性的调节。讨论了mRNA在癌细胞可塑性中的转录调节的作用,包括结合转录因子,DNA甲基化,组蛋白修饰和增强子。此外,辩论了mRNA编辑在癌细胞可塑性中的作用,包括mRNA剪接和mRNA修饰。此外,阐述了非编码(NC)RNA在癌症可塑性中的作用,包括microRNA,长基因间NCRNA和圆形RNA。最后,讨论了靶向癌细胞可塑性克服转移和癌症治疗性的不同策略。

基于模拟群落评估中欧鱼类环境 DNA 条形码的五对引物
环境 DNA (eDNA) 宏条形码已成为检查鱼类群落的有力工具。在将基于 eDNA 的评估引入监管监测环境(例如欧盟水框架指令)之前,需要方法标准化。为了确保方法的准确性并满足监管标准,已经建立了各种采样、实验室和生物信息学工作流程。然而,全面监测鱼类的关键先决条件是选择合适的引物对,以准确识别给定水体中存在的鱼类。过去十年中,发表了针对不同遗传标记区域的各种鱼类特异性引物对。然而,尚未开展专门研究来评估常用鱼类引物对在评估中欧鱼类物种方面的性能。因此,我们创建了一个由 45 种中欧鱼类 DNA 组成的人工“模拟”群落,并检查了五对引物的检测能力和可重复性。我们的研究重点介绍了引物选择和生物信息学过滤对 eDNA 宏条形码结果的影响。在我们研究中评估的五对引物中,tele02(12S 基因)引物对是中欧淡水鱼 eDNA 元条形码的最佳选择。此外,MiFish-U(12S)和 SeaD NA-mid(COI)引物对表现出良好的检测能力和可重复性。然而,特异性较低的引物对(即针对脊椎动物)被发现不太可靠,并产生大量假阳性和假阴性检测。我们的研究说明了如何通过精心选择引物对和生物信息学流程使 eDNA 元条形码成为鱼类监测更可靠的工具。

DNA元法估计的丰度估计 微生物中的作用在动物入侵中:范围评论 一个具有零排放的新过程,用于真正的生物降解... 一个具有零排放的新过程,用于真正的生物降解... 评估环境DNA的五个引物对...
作者格式,未经同行评审的文件发布于2023年7月7日。doi:https://doi.org/10.3897/arphapreprints.e109709