XiaoMi-AI文件搜索系统
World File Search SystemHifi

通过长读长重新审视非模型物种的基因组,可以对其生物学和进化产生新的见解
使用长读数据获得的高质量基因组不仅可以更好地了解杂合性水平、重复内容以及与使用短读技术获得的基因组相比更准确的基因注释和预测,而且还可以帮助了解单倍型分化。近年来,长读测序技术的进步使得为非模式生物生成此类高质量组装成为可能。这使我们能够重新审视基因组,而使用前几代数据和组装软件将其组装到染色体规模上一直存在问题。线虫是后生动物中种类最多、种类最多的动物门之一,但对其研究仍然很少,许多以前组装的基因组都是碎片化的。使用 Nanopore R10.4.1 和 PacBio HiFi 获得的长读长,我们生成了 Mermithidae 科二倍体线虫的高度连续组装体,目前尚未获得该科的密切相关基因组,以及 Panagrolaimidae 科三倍体线虫的折叠组装体和分阶段组装体。这两个基因组之前都已分析过,但碎片组装体的支架大小与组装前的长读长长度相当。我们的新组装体说明了长读长技术如何更好地表示物种基因组。我们现在能够根据更完整的基因和转座因子预测进行更准确的下游分析。

四倍体中国樱桃(Prunus pseudocerasus)的单倍型解析基因组组装为果实硬度提供了见解
中国樱桃(Prunus pseudocerasus)是中国主要的核果作物之一,具有十分重要的意义。然而,由于缺乏高质量的基因组资源,人工改良其性状和遗传分析具有挑战性,这主要归因于难以解析其四倍体和高度杂合的基因组。在此,我们使用 PacBio HiFi、Oxford Nanopore 和 Hi-C 组装了品种‘诸暨短柄饼’的染色体水平、单倍型解析基因组,包含 993.69 Mb,组装成 32 条假染色体。单倍型内比较分析揭示了广泛的基因组内序列和表达一致性。系统发育和比较基因组分析表明,P. pseudocerasus 是一个稳定的同源四倍体物种,与野生的 P. pusilliflora 密切相关,两者大约在 1834 万年前分化。与其他李属植物类似,樱桃也经历了一次常见的全基因组复制事件,该事件发生在大约 1.3996 亿年前。由于果实硬度低,樱桃不适合长距离运输,从而限制了其在中国的快速发展。在成熟果实阶段,樱桃品种‘诸暨短柄梨’的硬度明显低于樱桃品种‘黑珍珠’。硬度的差异归因于果胶、纤维素和半纤维素含量变化的程度。此外,比较转录组分析发现了两个参与果胶生物合成的基因 GalAK-like 和 Stv1,这可能是造成‘诸暨短柄梨’和‘黑珍珠’果实硬度差异的原因。PpsGalAK-like 和 PpsStv1 的瞬时转化会增加原果胶含量,从而提高果实硬度。我们的研究为中国樱桃功能基因组学研究和重要园艺性状的提升奠定了坚实的基础。

单倍型解析的染色体水平鳄梨基因组可用于分析新的鳄梨基因
鳄梨 (Persea americana) 是木兰科植物的一种,木兰科植物是被子植物的早期分支谱系,其果实营养丰富,在全球具有很高的价值。在这里,我们报告了商业鳄梨品种 Hass 的染色体水平基因组组装,该品种占世界鳄梨消费量的 80%。使用由遗传图谱支持的先前发布的基因组版本进一步组装由 Pacific Biosciences HiFi 读数产生的 DNA 重叠群。总组装体为 913 Mb,重叠群 N50 为 84 Mb。分配给 12 条染色体的重叠群代表 874 Mb,覆盖了 98.8% 的胚性植物基准单拷贝基因。蛋白质编码序列注释确定了 48 915 个鳄梨基因,其中 39 207 个可归因于功能。基因组含有 62.6% 的重复元素。研究了基因组中感兴趣的特定生物合成途径。分析表明,鳄梨中庚糖生物合成的主要途径可能是通过景天庚酮糖 1,7 双磷酸,而不是通过其他途径。内切葡聚糖酶基因数量众多,与鳄梨使用纤维素酶催熟果实一致。尽管经历了多次基因组复制事件,但鳄梨基因组似乎在同源染色体之间有有限数量的易位。与相关物种的蛋白质组聚类允许识别鳄梨和樟科其他成员特有的基因,以及在单子叶植物和真双子叶植物分化前或分化时分化的物种特有的基因。该基因组提供了一种工具,以支持未来开发产量和果实质量更高的优质鳄梨品种。

棕色野兔(Lepus Europaeus)的高质量基因组组装与染色体水平脚手架
我们在这里提出了棕色野兔(Lepus europaeus pallas)的高质量基因组组装,该组件基于来自芬兰东部利珀里(Liperi)的雄性标本的纤维细胞细胞系。这个棕色的野兔基因组代表了芬兰对欧洲参考基因组试验e ort e ort的第一个贡献,以生成欧洲生物多样性的参考基因组。使用HI-C染色体结构捕获方法,使用25倍PACBIO HIFI测序数据组装了基因组,并使用了SCA的旧基因组。在手动策划后,组装的基因组长度为2,930,972,003 bp,N50 sca egs为125.8 MB。93.16%的组装可以分配给25个识别的染色体(23个常染色体加X和Y),与已发布的核型匹配。染色体根据大小编号。基因组基于BUSCO分数(MAM-malia_odb10数据库)具有高度的完整性,完成:96.1%[单副本:93.1%,重复:3.0%],片段为0.8%,缺少2.9%。对细胞系的线粒体基因组进行测序并分别组装。最终注释的基因组具有30,833个基因,其中21,467个多肽代码。棕色野兔基因组特别有趣,因为该物种很容易与北部欧亚大陆物种接触区的山野兔(Lepus timidus L.)杂交,从而产生肥沃的春季,并导致这两个物种之间的基因流。除了为人群研究提供有用的比较外,基因组还可以深入了解一般的毛刺和lagomorpha之间的染色体演化。基因组的染色体组装还表明,细胞系在培养过程中尚未获得核型变化。

T2T参考基因组组装及全基因组关联研究揭示杨梅果实品质的遗传基础
杨梅 (Myrica rubra 或 Morella rubra;2n = 16) 所产果实风味独特、营养丰富、经济价值高。然而,先前版本的杨梅基因组缺乏序列连续性。此外,迄今为止,尚无大规模种质资源关联分析检查过决定果实品质性状的等位基因和遗传变异。因此,在本研究中,我们使用 PacBio HiFi 长读长为品种‘早嘉’组装了一个端粒到端粒 (T2T) 无间隙参考基因组。得到的 292.60 Mb T2T 基因组揭示了 8 个着丝粒区域、15 个端粒和 28 345 个基因。这代表着杨梅基因组的连续性和完整性的显著提高。随后,我们对 173 个种质进行了重新测序,鉴定出 6 649 674 个单核苷酸多态性 (SNP)。此外,29 个果实品质相关性状的表型分析促成了全基因组关联研究 (GWAS),该研究鉴定了与 28 种性状显著相关的 1937 个 SNP 和 1039 个基因。在 Chr6:3407532 至 5 153 151 bp 区域上鉴定了一个与果实颜色相关的 SNP 簇,包含两个 MYB 基因(MrChr6G07650 和 MrChr6G07660),这些基因在极端表型转录组中表现出差异表达,与花青素合成有关。一个相邻的、紧密连锁的基因 MrChr6G07670(MLP 样蛋白)包含一个外显子错义变体,经证实可使烟草叶片中的花青素产量增加十倍。这个 SNP 簇可能是一个数量性状基因座 (QTL),它共同调控杨梅果实的颜色。总之,我们的研究提出了一个完整的参考基因组,揭示了一系列与果实品质性状相关的等位基因变异,并确定了可以利用来提高杨梅果实品质和育种效率的功能基因。
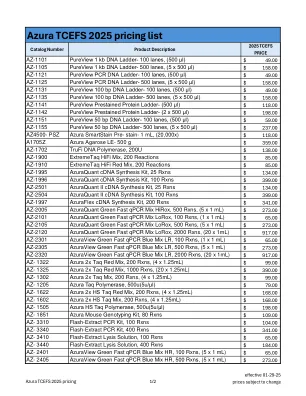
Azura TCEFS 2025 价格表
价格 AZ-1101 PureView 1 kb DNA 梯度 - 100 道, (500 μl) 49.00 美元 AZ-1105 PureView 1 kb DNA 梯度 - 500 道, (5 x 500 μl) 158.00 美元 AZ-1121 PureView PCR DNA 梯度 - 100 道, (500 μl) 49.00 美元 AZ-1125 PureView PCR DNA 梯度 - 500 道, (5 x 500 μl) 158.00 美元 AZ-1131 PureView 100 bp DNA 梯度 - 100 道, (500 μl) 49.00 美元 AZ-1135 PureView 100 bp DNA 梯度 - 500 道, (5 x 500 μl) AZ-1141 PureView 预染蛋白梯度(500 μl) AZ-1142 PureView 预染蛋白梯度(2 x 500 μl) AZ-1151 PureView 50 bp DNA 梯度(100 泳道,500 μl) AZ-1155 PureView 50 bp DNA 梯度(500 泳道,5 x 500 μl) AZ4500- PSZ Azura SmartStain 预染液(1 mL,20,000x) A1705Z Azura 琼脂糖 LE(500 g) AZ-1702 TruFi DNA 聚合酶,200U AZ-1900 ExtremeTaq HiFi Mix,200 次反应 AZ-1910 ExtremeTaq HiFi Red Mix,200 次反应 AZ-1995 AzuraQuant cDNA 合成试剂盒,25 次反应 AZ-1996 AzuraQuant cDNA 合成试剂盒,100 次反应 AZ-2501 AzuraQuant II cDNA 合成试剂盒,25 次反应 AZ-2504 AzuraQuant II cDNA 合成试剂盒,100 次反应 AZ-1997 AzuraFlex cDNA 合成试剂盒,200 次反应 AZ-2005 AzuraQuant Green Fast qPCR Mix HiRox,500 次反应,(5 x 1 毫升) 273.00 美元 AZ-2101 AzuraQuant Green Fast qPCR Mix LoRox,100 次反应,(1 x 1 毫升) 65.00 美元 AZ-2105 AzuraQuant Green Fast qPCR Mix LoRox,500 次反应,(5 x 1 毫升) 273.00 美元 AZ-2120 AzuraQuant Green Fast qPCR Mix LoRox,2000 次反应,(20 x 1 毫升) 917.00 美元 AZ-2301 AzuraView Green Fast qPCR Blue Mix LR,100 次反应,(1 x 1 毫升) 65.00 美元 AZ-2305 AzuraView Green Fast qPCR Blue Mix LR,500 次反应,(5 x 1 毫升) AZ-2320 AzuraView Green Fast qPCR Blue Mix LR,2000 次反应,(20 x 1 毫升) AZ-1322 Azura 2x Taq Red Mix,200 次反应,(4 x 1.25 毫升) AZ-1325 Azura 2x Taq Red Mix,1000 次反应,(20 x 1.25 毫升) AZ-1302 Azura 2x Taq Mix,200 次反应,(4 x 1.25 毫升) AZ-1205 Azura Taq 聚合酶,500u(5u/μl) AZ-1622 Azura 2x HS Taq Red Mix, 200 次反应,(4 x 1.25 毫升) 168.00 美元 AZ- 1602 Azura 2x HS Taq 混合液,200 次反应,(4 x 1.25 毫升) 168.00 美元 AZ- 1505 Azura HS Taq 聚合酶,500u (5u/μl) 198.00 美元 AZ- 1851 Azura 小鼠基因分型试剂盒,80 次反应 109.00 美元 AZ- 3310 Flash-Extract PCR 试剂盒,100 次反应 104.00 美元 AZ- 3340 Flash-Extract PCR 试剂盒,400 次反应 341.00 美元 AZ- 3410 Flash-Extract 裂解液,100 次反应 65.00 美元 AZ- 3440 Flash-Extract 裂解液,400 次反应 184.00 美元 AZ-2401 AzuraView Green Fast qPCR Blue Mix HR,100 次反应,(5 x 1 毫升) 65.00 美元 AZ-2405 AzuraView Green Fast qPCR Blue Mix HR,500 次反应,(5 x 1 毫升) 273.00 美元

封面页奖项 - 小麦库
1。摘要第3年:在项目的第三年中,小麦繁殖者发布了28种商业品种(7种带有PVP),并在Grin-Global中存放了5种改进的种系。由28个新的同行评审论文证明了小麦库参与者的良好生产率。该项目还生成了HIFI基因组数据,用于在五个组织中为200个辅助的10种小麦和表达数据的10种不同的表达数据。我们还使用了单分子鱼和单细胞转录组学中的定量多路复用,以鉴定发育中的小麦尖峰中的不同细胞群体,并生成小麦峰值发育的表达地图集。探索了几个基因分型平台,并使用实用的单倍型图V1进行了插补,从而集成了T3中存在的不同平台的标记。已经启动了改进的PHG V2的开发。从290次现场试验中收集了57,800个地块的农艺数据,并从119英亩以上的282次UAS航班收集了其他现场数据,其中包括35,376个地块。通过育种程序和基因分型实验室产生> 350 m的数据点,共有48,900个样品。此信息用于在公共小麦育种计划中实施基因组选择,也是宝贵的研究资源。在2024年,有8位学生成功完成了博士学位,另外50名参加了由项目教育协调员组织的多项教育活动,包括面对面的研讨会和在线活动。分别在附录1和2中分别提供已发布品种和出版物的完整列表。学生的个人资料和项目以及项目会议和教育资源的链接可在WheatCap网站(https://www.triticeaecap.org/)上获得。社区资源在附录3中,在附录4中接受培训的学生。附录5提出了基因分型实验室进行的调查结果。

染色体规模的基因组组装面包小麦的野生相对timopheevii 1 2 Surbhi Grewal 1,Cai-Yun Yang 1,Duncan Scholefield 1,S
Chromosome-scale genome assembly of bread wheat's wild relative Triticum timopheevii 1 2 Surbhi Grewal 1 , Cai-yun Yang 1 , Duncan Scholefield 1 , Stephen Ashling 1 , Sreya Ghosh 2 , David 3 Swarbreck 2 , Joanna Collins 3 , Eric Yao 4,5 , Taner Z. Sen 4,5 , Michael Wilson 6 , Levi Yant 6 , Ian P. King 1和4 Julie King 1 5 6 1。麦片研究中心,植物与作物科学系,生物科学学院,诺丁汉大学7号大学,拉夫堡,LE12 5rd,英国8 2。伯爵研究所,诺里奇研究公园,诺里奇NR4 7UZ,英国9 3。基因组参考信息学团队,惠康桑格学院,惠康信托基因组10校园,欣克斯顿,CB10 1RQ,英国11 4。加利福尼亚大学加利福尼亚大学,加利福尼亚州伯克利生物工程系,美国94720,美国12 5。 美国农业部 - 农业研究服务局,西部地区13研究中心,农作物改善与遗传学研究部门,布坎南街800 诺丁汉大学,大学公园,诺丁汉,NG7 2rd 16通讯作者:Surbhi Grewal(surbhi.grewal@nottingham.ac.uk)17 18摘要19 20 20小麦(Triticum aestivum)是最重要的食物作物之一,迫切需要增加生产的生产,以养活生长的世界。 triticum timopheevii(2n = 4x = 28)是一种同种二磷酸22小麦野生物种,其中包含在许多23个先前的小麦改善育种计划中利用的A T和G基因组。 在这项研究中,我们报告了基于PACBIO 25 HIFI读取和染色体构象捕获(HI-C)的24个染色体尺度参考基因组组装PI 94760。 ex asch。加利福尼亚大学加利福尼亚大学,加利福尼亚州伯克利生物工程系,美国94720,美国12 5。美国农业部 - 农业研究服务局,西部地区13研究中心,农作物改善与遗传学研究部门,布坎南街800诺丁汉大学,大学公园,诺丁汉,NG7 2rd 16通讯作者:Surbhi Grewal(surbhi.grewal@nottingham.ac.uk)17 18摘要19 20 20小麦(Triticum aestivum)是最重要的食物作物之一,迫切需要增加生产的生产,以养活生长的世界。 triticum timopheevii(2n = 4x = 28)是一种同种二磷酸22小麦野生物种,其中包含在许多23个先前的小麦改善育种计划中利用的A T和G基因组。 在这项研究中,我们报告了基于PACBIO 25 HIFI读取和染色体构象捕获(HI-C)的24个染色体尺度参考基因组组装PI 94760。 ex asch。诺丁汉大学,大学公园,诺丁汉,NG7 2rd 16通讯作者:Surbhi Grewal(surbhi.grewal@nottingham.ac.uk)17 18摘要19 20 20小麦(Triticum aestivum)是最重要的食物作物之一,迫切需要增加生产的生产,以养活生长的世界。triticum timopheevii(2n = 4x = 28)是一种同种二磷酸22小麦野生物种,其中包含在许多23个先前的小麦改善育种计划中利用的A T和G基因组。在这项研究中,我们报告了基于PACBIO 25 HIFI读取和染色体构象捕获(HI-C)的24个染色体尺度参考基因组组装PI 94760。ex asch。组件的总尺寸为26 9.35 GB,具有42.4 Mb的重叠元素N50和166,325个预测的基因模型。DNA甲基化27分析表明,G基因组的平均甲基化碱基比A T基因组更多。28 g基因组也与aegilops speltoides的S基因组更紧密相关,而不是与六倍体或四倍体小麦的B 29基因组。总而言之,T。timopheevii基因组组装为30发现了对食品31安全性的农艺重要基因的基因组发现的宝贵资源。32 33背景和摘要34 35人物属包括许多野生和栽培的小麦种类,包括二倍体,四倍体36和六倍体形式。多倍体物种起源于甲状腺素和37个相邻的Aegilops属(山羊草)之间的杂交。四倍体物种,毛triticum triticum tricum torgidum(2n = 4x = 28,38 aabb),也称为emmer小麦,三质体timopheevii(2n = 4x = 4x = 28,a t a t gg)是39多态的。triticum urartu thum。ex gandil(2n = 2x = 14,aa)是这两个物种1的基因组供体1,而B和G基因组与Aegilops 41 Speltoides 2的S基因组密切相关。两种四倍体物种均具有野生和驯化的形式,即T. turgidum L. ssp。42 dicoccoides(Körn。&graebn。)Thell。和SSP。dicoccum(schrankexschübl。)thell。,分别为43,T。Timopheevii(Zhuk。)Zhuk。 ssp。 armeniacum(jakubz。) slageren和ssp。 分别为44 timopheevii。 durum(desf。) 45 HUSN。Zhuk。ssp。armeniacum(jakubz。)slageren和ssp。分别为44 timopheevii。durum(desf。)45 HUSN。45 HUSN。此外,四倍体硬质小麦T. turgidum L. ssp。(2n = 4x = 28,AABB),用于意大利面的生产,六倍层面包小麦triticum aestivum aestivum 46 L.(2n = 6x = 42,aabbdd)从驯养的emmer小麦中进化而成,后者与aegilops tauschii(d tauschii donore hybridations the the the the the bentertiationally the tauschii donore(d genuschii donor)(d donore)6,000,000,000,000,000,000。十六世纪48个Triticum Zhukovskyi(Aagga M a M)源自培养的Timopheevii杂交和49个培养的Einkorn triticum单球菌3(2n = 2x = 2x = 14,A M A M)。50 51

与长期水族馆设施中的热带八放珊瑚相关的四种新型 Endozioicomonas 菌株的基因组序列
我们报告了从葡萄牙里斯本海洋馆 19 立方米热带展览水族馆中保存的两个 Litophy ton sp. 标本中分离出的四种 Endozoicomonas 菌株的基因组。如前所述 (2) 回收宿主衍生的微生物细胞悬浮液。将一克珊瑚组织在 9 mL 无菌 Ca 2+ - 和 Mg 2+ - 人工海水中均质化 (2)。将匀浆连续稀释,分别接种在 1:2 稀释的海洋琼脂和 1:10 稀释的 R2A 培养基上,并在 21°C 下孵育 4 周。使用 Wizard 基因组 DNA 纯化试剂盒 (Promega, USA) 从 1:2 海洋肉汤中新鲜生长的培养物中提取单个菌落的基因组 DNA。使用通用引物 (F27 和 R1492) 从基因组 DNA 中扩增 16S rRNA 基因,通过 Sanger 测序来确认纯度。使用 SILVA 比对、分类和树服务 (v1.2.12) 和数据库 (v138.1) 进行分类分配。使用 PacBio 测序技术 (5),相同的基因组 DNA 样本在 DOE 联合基因组研究所 (JGI) 进行基因组测序。对于每个样本,将基因组 DNA 剪切至 6-10 kb,使用 SMRTbell Express Template Prep Kit 3.0 进行处理,并用 SMRTbell 清理珠 (PacBio) 进行纯化。使用条形码扩增寡核苷酸 (IDT) 和 SMRTbell gDNA 样本扩增试剂盒 (PacBio) 富集纯化产物。构建了 10 kb PacBio SMRTbell 文库,并使用 HiFi 化学在 PacBio Revio 系统上进行测序。使用 BBTools v.38.86 ( http://bbtools.jgi.doe.gov ) 根据 JGI 标准操作规范 (SOP) 协议 1061 对原始读段进行质量过滤。使用 Flye v2.8.3 (6) 组装过滤后的 >5 kb 读段。生物体和项目元数据存放在 Genomes OnLine 数据库中 (7)。使用 NCBI 原核基因组注释流程 (PGAP v.6.7) (8) 和 DOE-JGI 微生物基因组注释流程 (MGAP v.4) (9) 对重叠群进行注释,并与集成微生物基因组和微生物组系统 v7 (IMG/M) 相结合进行比较分析 (10)。使用 CheckM 评估基因组完整性和污染

与长期水族馆设施中的热带八放珊瑚相关的四种新型 Endozioicomonas 菌株的基因组序列
我们报告了从葡萄牙里斯本海洋馆 19 立方米热带展览水族馆中保存的两个 Litophy ton sp. 标本中分离出的四种 Endozoicomonas 菌株的基因组。如前所述 (2) 回收宿主衍生的微生物细胞悬浮液。将一克珊瑚组织在 9 mL 无菌 Ca 2+ - 和 Mg 2+ - 人工海水中均质化 (2)。将匀浆连续稀释,分别接种在 1:2 稀释的海洋琼脂和 1:10 稀释的 R2A 培养基上,并在 21°C 下孵育 4 周。使用 Wizard 基因组 DNA 纯化试剂盒 (Promega, USA) 从 1:2 海洋肉汤中新鲜生长的培养物中提取单个菌落的基因组 DNA。使用通用引物 (F27 和 R1492) 从基因组 DNA 中扩增 16S rRNA 基因,通过 Sanger 测序来确认纯度。使用 SILVA 比对、分类和树服务 (v1.2.12) 和数据库 (v138.1) 进行分类分配。使用 PacBio 测序技术 (5),相同的基因组 DNA 样本在 DOE 联合基因组研究所 (JGI) 进行基因组测序。对于每个样本,将基因组 DNA 剪切至 6-10 kb,使用 SMRTbell Express Template Prep Kit 3.0 进行处理,并用 SMRTbell 清理珠 (PacBio) 进行纯化。使用条形码扩增寡核苷酸 (IDT) 和 SMRTbell gDNA 样本扩增试剂盒 (PacBio) 富集纯化产物。构建了 10 kb PacBio SMRTbell 文库,并使用 HiFi 化学在 PacBio Revio 系统上进行测序。使用 BBTools v.38.86 ( http://bbtools.jgi.doe.gov ) 根据 JGI 标准操作规范 (SOP) 协议 1061 对原始读段进行质量过滤。使用 Flye v2.8.3 (6) 组装过滤后的 >5 kb 读段。生物体和项目元数据存放在 Genomes OnLine 数据库中 (7)。使用 NCBI 原核基因组注释流程 (PGAP v.6.7) (8) 和 DOE-JGI 微生物基因组注释流程 (MGAP v.4) (9) 对重叠群进行注释,并与集成微生物基因组和微生物组系统 v7 (IMG/M) 相结合进行比较分析 (10)。使用 CheckM 评估基因组完整性和污染