XiaoMi-AI文件搜索系统
World File Search SystemMarie
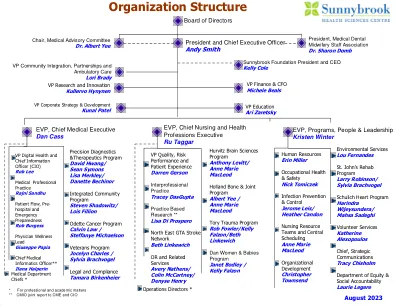
组织结构 - 多伦多
Operations Directors * - Hurvitz Brain Sciences Anne Marie MacLeod - Integrated Community Program Lois Fillion - Odette Cancer Steffanye Michaelson - St. John s Rehab Sylvia Brachvogel - Veterans Sylvia Brachvogel - Schulich Heart Mahsa Sadeghi - Holland Bone & Joint Anne Marie MacLeod - Tory Trauma Kelly Falzon/Beth Linkewich/Rob福勒 - 妇女和婴儿凯利·法尔松(Kelly Falzon) - 或与相关服务拒绝亨利(Henry) - 精选诊断和治疗学Lisa Merkley/ Danette Beechinor div>

活动计划 - 9月。 11。2024 -GOSEC会议
控制与第三方相关的风险:建立适合您组织的控制和监视环境Marie -eve Girard&Catherine Boivin -Me.Cie&Richter

绿色AI:人工智能和能源革命的政策议程
2024年5月Kenddrick Chan Devorah West Marie Teo Harriet Harriet Brown Tom Westgarth Thomas Smith

维生素D状况在怀孕的头三个月与妊娠糖尿病的关系 - 嵌套病例 - 控制研究
Eleanor Salakos,Tioka Rabeony,Marie Courbaisse,JoëlleTaieb,Vassilis Tsatsaris和Al。临床营养,2021,40,pp.79-86。10.1016/j.clnu.2020.04.04.028。

人类结膜器官研究眼表稳态和疾病
玛丽·弗拉姆(Marie Flag-He)在狮子,1,2, * jeroen korving,1,1,1 19 5 Yawata nobuyo,6,7,7 Yawata,9,9,10,10,12,12,13,14 L.S.
人类 - 界面 - 甲状腺素至研究的 - 眼曲面 -
玛丽·弗拉姆(Marie Flag-He)在狮子,1,2, * jeroen korving,1,1,1 19 5 Yawata nobuyo,6,7,7 Yawata,9,9,10,10,12,12,13,14 L.S.

劳动期间使用催产素的最佳实践实施策略
Rebecca Marie Falzon 1,3,Stephen Falzon 3,Yves Muscat Baron 2,Lilian M Azzopardi 1 1 1 1药学系,马耳他马耳他大学医学与外科学院,

单核细胞人白细胞抗原DR表达患有心肺旁路的年轻婴儿
Alexis Chenouard,Marie Rimbert,Nicolas Joram,CécileBraudeau,Antoine Roquilly等。胸腔手术年鉴,2021,111(5),第1636-1642页。10.1016/j.athoracsur.2020.05.071。hal-04706563
