XiaoMi-AI文件搜索系统
World File Search SystemNextFlow

CRISPR-Analytics(CRISPR-A):基因编辑精确分析和模拟的平台
摘要 使用当前可用的工具进行基因编辑表征并不总是能给出编辑大量细胞中存在的不同类型基因编辑之间的精确相对比例。我们开发了 CRISPR-Analytics,即 CRISPR-A,它是一种全面而多功能的基因组编辑 Web 应用工具和 nextflow 流程,用于支持基因编辑实验设计和分析。CRISPR-A 提供了一个由数据分析工具和模拟组成的强大的基因编辑分析流程。它比当前工具实现了更高的精度并扩展了功能。分析包括基于模拟的噪声校正、spike-in 校准扩增偏差减少和高级交互式图形。这种扩展的稳健性使该工具成为分析高度敏感的案例(例如临床样本或编辑效率低的实验)的理想选择。它还通过模拟基因编辑结果提供对实验设计的评估。因此,CRISPR-A 非常适合支持多种实验,例如基于双链 DNA 断裂的工程、碱基编辑 (BE)、引物编辑 (PE) 和同源定向修复 (HDR),而无需指定使用的实验方法。

balrog-mon:用于基因组牛津纳米孔
宏基因组测序是一种最近可行的方法,可以同时表征样品中的ARG,微生物组和病原体的数据,与分离和培养细菌相比,它是一种更有效,更全面的方法。对宏基因组数据的典型分析涉及一种基于组装的方法或基于读取的方法,每种方法都有其自身的好处和限制。宏基因组装配允许对ARGS进行上游或下游研究,并提供对其起源的准确识别。但是,这种方法可能导致信息丢失,因为低覆盖的基因组通常不会组装。相比之下,基于读取的方法可实现所有可用数据的映射,但缺乏探索周围基因组环境或提供准确分类分类的能力。为了应对这些挑战,我们开发了Balrog-mon,这是一种多功能且可重现的NextFlow管道,用于测量病原体和元基因组长阅读测序的ARG,提供“组装”和“无装配”工作流程选项。

使用长阅读的RNA测序推进基因组注释:Igdrion设施的见解
1。van Dijk El,Jaszczyszyn Y,Naquin D,ThermesC。测序技术的第三次革命是测序技术的简短历史。遗传学趋势2018; 34:666–81。 2。 di Tommaso P,Chatzou M,Floden EW,Barja PP,Palumbo E,NotredameC。NextFlow启用可重现的计算工作流程。 NAT Biotechnol 2017; 35:316–9。 3。 Chen Y,Sim A,Wan YK,Yeo K,Lee JJX,Ling MH等。 与BAMBU的长阅读RNA-seq数据中的上下文感知的转录本定量。 NAT方法2023; 20:1187–95。 4。 Wucher V,Legeai F,HédanB,Rizk G,Lagoutte L,Leeb T等。 feefnc:长期非编码RNA注释及其应用于狗转录组的工具。 核酸研究2017; 45:1-12。遗传学趋势2018; 34:666–81。2。di Tommaso P,Chatzou M,Floden EW,Barja PP,Palumbo E,NotredameC。NextFlow启用可重现的计算工作流程。NAT Biotechnol 2017; 35:316–9。3。Chen Y,Sim A,Wan YK,Yeo K,Lee JJX,Ling MH等。 与BAMBU的长阅读RNA-seq数据中的上下文感知的转录本定量。 NAT方法2023; 20:1187–95。 4。 Wucher V,Legeai F,HédanB,Rizk G,Lagoutte L,Leeb T等。 feefnc:长期非编码RNA注释及其应用于狗转录组的工具。 核酸研究2017; 45:1-12。Chen Y,Sim A,Wan YK,Yeo K,Lee JJX,Ling MH等。与BAMBU的长阅读RNA-seq数据中的上下文感知的转录本定量。NAT方法2023; 20:1187–95。4。Wucher V,Legeai F,HédanB,Rizk G,Lagoutte L,Leeb T等。feefnc:长期非编码RNA注释及其应用于狗转录组的工具。核酸研究2017; 45:1-12。

Samuel Bharti
•编程和脚本:熟练掌握R,Python,Matlab,Bash进行数据分析,统计建模和生物信息学管道。•生物信息学:在单细胞和空间转录组学,变体分析,RNA-seq和多摩学集成中经验丰富。•数据分析和可视化:使用Seurat,Deseq2和Pseudobulk等工具开发自定义工作流程进行生物数据分析的熟练。使用GGPLOT2和绘图的数据可视化中的强大功能。•云和高性能计算:设置和管理云基础架构(AWS,GCP)和HPC环境方面的专业知识,使用Slurm和Docker进行可扩展计算。•软件和Web开发:开发了带有r闪亮,简化和反应的生物信息学Web应用程序,重点是交互式数据探索。•机器学习与建模:应用机器学习技术到生物医学数据,具有特征选择,分类模型和网络分析的经验。•工作流程管理:使用NextFlow和管道开发进行大规模基因组数据处理的工作流管理经验。

genepi:用于整个基因组测序分析的GPU增强的下一代生物信息学管道
驱动了对高级计算基础架构进行分析这些大数据集的需求。这项工作的目的是引入一条创新的生物信息学管道,名为Genepi,以进行WGS简短配对读数的有效和精确分析。构建在具有模块化结构的NextFlow框架上,Genepi结合了GPU加速算法并支持多种工作流程配置。管道可自动从生物学WGS数据中提取生物学相关的见解,包括:与疾病相关的变体,例如单核苷酸变体(SNV),小插入或缺失(Indels),拷贝数变体(CNV)和结构变体(SVS)。针对高性能计算(HPC)环境进行了优化,它利用了工作 - 安排的提交,并行处理以及为每个分析步骤量身定制的资源分配。对合成数据集进行了测试,Genepi准确地识别了基因组变量,并且具有与最新工具相当的性能。这些功能使Genepi成为研究和临床环境中大规模分析的宝贵工具,这是朝着建立国家计算和技术医学中心的关键一步。

CRISPR-Analytics(CRISPR-A):用于基因编辑的精确分析和模拟平台
并不总是在编辑的大部分细胞中存在的不同类型的基因编辑中提供精确的相关比例。我们已经开发了CRISPR-Analytics,CRISPR-A,它是一种全面且通用的基因组编辑Web应用程序工具和NextFlow Pipeline,可为基因编辑实验设计和分析提供支持。CRISPR-A提供了由数据分析工具和仿真组成的强大基因编辑分析管道。它的准确性比Curlant工具更高,并扩展了功能。分析包括基于模拟的噪声校正,升高的校准放大偏置降低和高级交互式图形。这种扩展的鲁棒性使该工具非常适合分析高度敏感的病例,例如临床样本或较低编辑效率的实验。它还通过模拟基因编辑结果来评估实验设计。因此,CRISPR-A是支持多种实验的理想选择,例如双链DNA断裂工程,基础编辑(BE),引物编辑(PE)和同源性修复(HDR),而无需指定使用的实验方法。
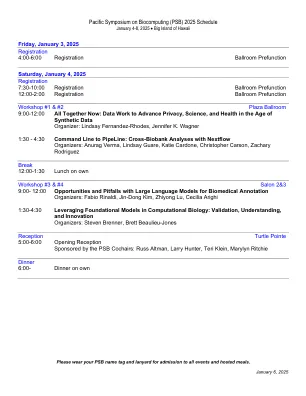
生物计算的太平洋研讨会(PSB)2025时间表
2025年1月4日,星期六,注册7:30-10:00注册宴会厅预选12:00-2:00注册宴会厅预选研讨会#1&#2 Plaza Ballroom 9:00-12:00现在在一起:数据工作:数据工作以促进隐私,科学,科学和健康的数据,瓦格纳(Wagner)1:30-4:30到管道的指挥行:与Nextflow组织者进行跨双obank分析:Anurag Verma,Lindsay Guare,Katie Cardone,Katie Carone,Christopher Carson,Zachary Rodriguez休息12:00-1:30,在自己的工作室#3&#4 Salon 2&3 9:00-12:00的午餐午餐中 Rinaldi, Jin-Dong Kim, Zhiyong Lu, Cecilia Arighi 1:30-4:30 Leveraging Foundational Models in Computational Biology: Validation, Understanding, and Innovation Organizers: Steven Brenner, Brett Beaulieu-Jones Reception Turtle Pointe 5:00-6:00 Opening Reception Sponsored by the PSB Cochairs: Russ Altman, Larry Hunter, Teri Klein,玛丽琳·里奇(Marylyn Ritchie)晚餐6:00-在自己的晚餐

CATH 2024:Cath-Alphaflow在CATH中的结构数量增加了一倍,并揭示了近200个新折叠 倒下的Apple检测作为辅助任务-UCL Discovery 使用环状伏安法的壳聚糖/还原石墨烯氧化石墨烯/锰二氧化碳改性电极检测胆固醇 药物交付科学与技术杂志 通过原位转化的脑室区域神经干细胞的原位转化
CATH(https://www.cathdb.info)从PDB中的实验蛋白结构和Alphafold数据库(AFDB)中预测的结构中分类的域结构。为了应对预测数据的规模,已经开发出一种新的NextFlow工作流量(Cath-Alphaflow),以将高质量的域分类为CATA超家族,并识别新颖的折叠组和超家族。Cath-Alphaflow使用一种新型的基于结构的结构域边界预测方法(Chainsaw)来识别多域蛋白质中的域。我们将CATA-AlphaFlow应用于未在21种模型生物体中的CATH和AFDB结构中分类的PDB结构,使CATH扩大了100%以上。域用于播种新颖的折叠,从PDB结构(2023年9月发行)中提供253个新折叠,而来自21个模型器官的蛋白质组织的AFDB结构中有96个。在可能的情况下,使用(i)从AFDB/uniprot50中的结构亲戚的注释中获得(i)预测(i)预测功能注释。我们还预测了功能部位和高度保守的残基。有些折叠与重要功能有关,例如光合作用的适应(感染植物),铁粘酶活性(在真菌中)和产后精子发生(在小鼠中)。Cath-Alphaflow将使我们能够在AFDB中识别更多的天主关系,从而进一步构成蛋白质结构景观。2024作者。由Elsevier Ltd.这是CC下的开放式访问文章(http://creativecom- mons.org/licenses/4.0/)。

使用机器学习在仓库机器人中优化自动采摘系统
许多领域的科学家,包括基因组学,材料科学和遥感,需要分析越来越多的数据[1、4、8、9]。科学工作流程系统促进了此类分析的自动化,使科学家能够从黑框任务中构成管道,并具有数据依赖性。由于这些工作流程通常用于处理大量数据,因此它们往往是资源密集型和长期运行的,从而导致大量的能源消耗,因此会导致碳排放。此外,大数据应用程序的日益普及已被确定为ICT行业排放量不断增加的驱动力[5]。因此,量化和理解科学工作流的碳足迹至关重要。诸如NextFlow [2]之类的科学工作流系统,允许在异质群中进行工作流程,执行和监视。尽管这些系统通常为执行的工作流程生成详细的性能跟踪和日志,但它们不会产生消耗或碳发射的能量记录。因此,用户必须用硬件/软件电表手动监视功耗,否则使用诸如Cloud Car-Bon Footprint(CCF)1或绿色算法(GA)[7]之类的方法,该方法采用线性功率模型将资源利用转化为能量消耗。在任何一种情况下,要将消耗的能量转化为发射的碳,用户需要一定量的碳强度(CI),例如年平均值或更细粒度的度量。没有此步骤,只能基于粗粒度资源利用度量来估算功耗。一般而言,CI测量每千瓦时消耗的电力(每千瓦时)产生的碳量(𝐶𝑂2)的量,并且根据产生电力的来源以及对电网的需求而在不同位置,季节和时间之间有所不同。实际上,监视功耗要求用户附加物理功率计或在执行工作流程之前启用基于软件的工具,例如英特尔的运行平均电源限制(RAPL)。这是可以使用CCF和GA工具的,但仅以降低的精度。两种方法都假定能源消耗线性缩放,这不一定在实践中存在[6]。更重要的是,为了构建线性功率模型,GA方法依赖于指定的计算资源的供应商指定的热设计功率(TDP),这是一个不反映关键处理器设置(例如处理器频率)的专有度量,并不表示IDLE功能消耗。此外,尽管两种方法都将功耗转移到碳排放中,但它们使用静态平均值来表示计算工作量消耗的电力CI,而忽略了CI通常是高度可变的。