XiaoMi-AI文件搜索系统
World File Search SystemNucleotide

数学模型用于研究遗传变异的限制性核酸内切酶
它。因此,如果像mtDNA这样的圆形DNA具有m识别(限制)位点,则该酶在消化后将其分散成M段。限制位点的数量和位置随核苷酸序列而变化。相比,两个DNA序列的相似性越高,裂解模式越接近。因此,可以通过比较限制位点的位置来估计两个同源DNA之间的核苷酸取代的数量。同样,可以从两个或分类的DNA片段的比例中估算核苷酸取代的数量。Upholt(8)研究了这两个问题,但他的锻炼并不一般,似乎涉及一些错误。fur-hoverore,upholt不关注种群中DNA序列的异质性明显高度(5)。当研究紧密相关的物种之间的遗传差异时,有必要消除这种异质性的作用。本文的目的是开发一个更严格的DNA遗传差异数学模型,并提出了一种统计方法,用于分析限制酶研究的数据。在前四个部分中,我们要么假设人群中没有多态性,要么仅考虑一对生物(个体)之间的遗传差异。在第五部分中将删除无多态性的假设。




基准生物学方法杂志| 2023 |卷。 10 | e99010003 doi:10.14440/jbm.2023.376基准组件免费纳米孔阅读映射
千足片是将叶子回收到热带生态系统中的土壤中的关键参与者。为了阐明其肠道菌群,我们从波多黎各的不同城市收集了千足虫。在这里,我们的目标是基准哪种方法最适合这个高度复杂的千足型微生物组的元基因组脱脂。我们用牛津纳米孔技术(ONT)奴才序列对肠道DNA进行了测序,然后使用Megan-LR,Kraken2蛋白模式,Kraken2核苷酸模式,GraphMap和MiniMAP2分析了数据,以对这些较长的ONT进行分类。从我们的两个样本中,我们分别获得了87,110和99,749个ONT读数。kraken2核苷酸模式与门和类分类级别的所有其他方法相比,读取最多的读取性,对两个样本中的读取中的75%进行了分类,其他方法未能分配足够的读数,以在类似物稀有曲线中产生分类曲线,以表明它们需要对这些进行分类的较大分类,以使这些曲线分为稀有曲线,以完全进行分类以进行分类。社区的各种方法是多种多样的,所有方法将两个样本中的20-50门分类。使用的读取和门类似于五个基准测试的读数和门的明显重叠。我们的结果表明,Kraken2核苷酸模式是应用这个高度复杂群落的宏基因组学脱脂的最合适工具。

MEGA12:分子进化遗传分析第 12 版
选择最合适的替换模型通常是分子系统发育学的初始步骤。模型选择的 ML 方法最初在 MEGA5(Tamura 等人,2011)中引入,并经常使用(补充图 S1)。MEGA 评估了六种主要核苷酸替换模型以确定最佳模型:通用时间可逆 (GTR)、Hasegawa-Kishino-Yano (HKY)、Tamura-Nei (TN93)、Tamura 3 参数 (T92)、Kimura 2 参数 (K2P) 和 Jukes-Cantor (JC);有关综述,请参阅(Nei and Kumar,2000)。这些主要替换模型描述了单个位点处核苷酸替换的瞬时概率。它们可以与位点间速率变化的(离散化)Gamma 分布(用 +G 表示)和不变位点的存在/不存在(用 +I 表示)相结合,这些模型在 Nei 和 Kumar(2000)中进行了综述。

ASRC-纽约城市大学
星期三,2025 年 1 月 29 日 咖啡 11:30 AM 研讨会 12:00-1:00 PM Riccardo Miggiano 药学副教授 意大利诺瓦拉东皮埃蒙特大学 David Jeruzalmi 化学与生物化学教授 纽约城市大学 细菌核苷酸切除修复途径处理受损 DNA 的机制 摘要 核苷酸切除修复 (NER) 途径是细菌中最重要的 DNA 修复系统之一。UvrABC 核酸外切酶复合物由 UvrA、UvrB 和 UvrC 蛋白组成,构成了负责检测和去除 DNA 损伤的途径。这种多步骤机制需要蛋白质复合物的动态组装,并且依赖于 ATP 结合和水解。具体而言,UvrA 和 UvrB 蛋白对 DNA 进行初步检测以查找损伤,同时避开天然 DNA。
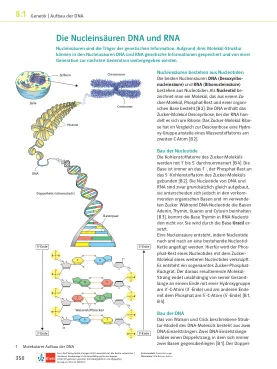
核酸DNA和RNA
核苷酸的构造糖分子的碳原子在1'至5英寸处编号[B 4]。碱始终与1'-,磷酸盐残基与糖分子的5´碳原子结合[B 2]。DNA和RNA的核苷酸通常是结构的,但是它们在前面的有机碱和糖的使用方面有所不同。虽然DNA-核苷酸含有腺苷,胸腺嘧啶,鸟嘌呤和胞嘧啶[B 3],但碱胸腺氨酸在RNA核酸中不发生。是由尿嘧啶基础制成的。核酸是通过逐渐将核苷酸添加到现有核苷酸链中而产生的。为此,核苷酸的磷酸盐其余部分与另一种核苷酸的糖分子有关。创建了所谓的糖磷酸骨链。所产生的分子链末端,无论其在一端的总长度如何,在3´-c原子(3´End)上的羟基和另一端,在5´-c原子(5´-end)上的磷酸盐[b 1,b 4]。

利用机器学习发现新型 SOS1 抑制剂
多种人类癌症的发病机制。1值得注意的是,KRAS 是一种常见突变,导致许多癌症病例中该基因的激活,包括 80% 至 90% 的胰腺癌、40% 至 50% 的结直肠癌和 30% 的非小细胞肺癌。1然而,对于携带 KRAS 突变的个体,临床治疗选择受到相当大的限制。目前,FDA 仅批准两种小分子抑制剂 sotorasib 和 adagrasib 用于治疗 KRAS G12C 突变的非小细胞肺癌,这表明 KRAS 靶向治疗的临床需求大大未得到满足。2,3 如图 1 所示,KRAS 的突变与 MAPK 家族中多种下游信号通路的激活有关,特别是 RAF – MEK – ERK 通路,它们对调节细胞存活和增殖至关重要。 1,4 RAS 蛋白起着分子开关的作用,在与鸟苷三磷酸 (GTP) 结合时处于活性开启状态,与与鸟苷二磷酸 (GDP) 结合时处于非活性关闭状态。5 这种开关受鸟苷酸交换因子和 GTPase 活化蛋白的调节,鸟苷酸交换因子促进 GDP 与 GTP 的交换,GTPase 活化蛋白增强 GTP 水解为 GDP。2 作为主要的鸟苷酸交换因子,Son of sevenless 1 (SOS1) 在 RAS 信号通路中起着至关重要的作用,它促进鸟苷酸交换并调节 KRAS 从“GDP 结合关闭状态”切换到“GTP 结合关闭状态”。

如何引用本文:Gallego-Munevar C,Carrillo-Godoy N,Rondón-Barragán是。 TPO基因相关的新型突变的分子检测
目的:这项研究的目的是分析来自诊断为先天性甲状腺功能减退症(CH)的CAT的甲状腺过氧酶(TPO)基因的不同片段的序列。材料和方法:由于您的流血刺激激素和低T4的血清浓度高,因此被诊断为猫科动物。从具有CH的狗的TPO基因中含有突变的序列的分析允许预测受影响CAT中基因中的突变位点。此外,基于聚合酶链反应测试的设计还可以放大和测序这些基因段。此外,在患者死亡后,进行了死灵病和组织病理学,寻找受影响器官的宏观和微观改变。结果:尸检检查表明甲状腺的心脏同心左心室高奖杯和甲状腺的双侧增大。甲状腺的组织病理学表现出卵泡性发育不全和低胶体产生。gDNA分析允许检测TPO基因中的突变,该突变与位于核苷酸14.627(G/A)中的核苷酸12.542(a> g)中的一个过渡相对应,在核苷酸和核苷酸30.713(g/c)中。结论:由于存在这些多态性,因此怀疑存在一种突变等位基因的单相表达。需要进行更多的研究,以了解杂合中杂合中的作用,以及与CH在CAT中相关的基因突变的作用。另一方面,本研究的数据是开发分子测试的基础,该测试可以快速准确诊断猫中的HC。